REPORTE DE CASO
PRESENTACIÓN ATÍPICA DE LA POLIANGEÍTIS MICROSCÓPICA CON COMPROMISO RENAL, PULMONAR, DERMATOLÓGICO Y DEL SISTEMA NERVIOSO CENTRAL
Fecha recibido: octubre 13 de 2017
Fecha aceptado: noviembre 17 de 2017
Rafael Parra-Medina MDa, Juliana Echeverri MDb, José Fernando Polo MDc, Jorge Alberto Carrillo MDd
a,c Servicio de Patología, Hospital de San José., Fundación Universitaria de Ciencias de la Salud. Bogotá D.C., Colombia.
b,d Servicio de Radiología., Hospital de San José., Fundación Universitaria de Ciencias de la Salud. Bogotá D.C., Colombia.
Resumen
La poliangeítis microscópica (PAM) es una vasculitis autoinmune caracterizada por la inflamación de los vasos sanguíneos de pequeño calibre y la presencia de anticuerpos anticitoplasma de neutrófilos (ANCA). Clínicamente se caracteriza por presentar glomerulonefritis y capilaritis pulmonar aunque la afectación de la piel, los nervios y el tracto gastrointestinal no es infrecuente. Presentamos el caso de un paciente de 17 años con manifestaciones atípicas dadas por glomerulonefritis pauci-inmune, hemorragia pulmonar, vasculitis leucocitoclásica cutánea y hemorragia cerebral. Además revisamos las características histológicas y radiológicas.
Palabras clave: poliangeítis microscópica, ANCA, glomerulonefritis, hemorragia pulmonar, vasculitis leucocitoclásica cutánea, hemorragia cerebral.
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS. This is an open access article under the CC BY-NC-ND license ( ñhttp://creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
Microscopic polyangiitis (MPA) is an autoimmune antineutrophil cytoplasmic antibody-associated (ANCA) small-vessel vasculitis. It is clinically characterized by glomerulonephritis and pulmonary capillaritis, however, skin, nerve and gastrointestinal tract involvement is also relatively common. A case is presented in a 17-year-old male patient with atypical manifestations, such as, pauci-immune glomerulonephritis, alveolar hemorrhage, cutaneous leukocytoclastic vasculitis and cerebral hemorrhage. MPA histologic and radiologic features were also reviewed.
Key words: Microscopic polyangiitis, ANCA, glomerulonephritis, pulmonary hemorrhage, cutaneous leukocytoclastic vasculitis, cerebral hemorrhage
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS. This is an open access article under the CC BY-NC-ND license ( ñhttp://creativecommons.org/licenses/by-nc-nd/4.0/).
Introducción
La polingeítis microscópica (PAM) es una vasculitis autoinmune categorizada dentro del grupo de las asociadas con anticuerpos anticitoplasma de neutrófilos (ANCA) positivos, al igual que el síndrome de Churg-Strauss, la vasculitis limitada al riñón y la granulomatosis de Wegener1, esta última denominada en la actualidad como granulomatosis con poliangeítis.2 La ultima clasificación dada por “Chapel Hill Consensus Conference Nomenclature of Vasculitides” ha definido la PAM como una vasculitis necrosante con mínimos o ausencia de depósitos inmunes. Esta entidad afecta en especial pequeños vasos como capilares, vénulas o arteriolas, aunque también puede comprometer arterias pequeñas y medianas.1
La PAM fue descrita en 1923 por Wohlwill y revisada por Davson y col en 1948. Se reconoció como una glomerulonefritis rápidamente progresiva con capilaritis pulmonar.3 La incidencia y la prevalencia de la enfermedad no es conocida, sin embargo se ha observado que afecta más a los pacientes del sur de Europa y Asia, mientras que la poliangeítis granulomatosa tiene mayor prevalencia en la población caucásica del norte de Europa y Norteamérica.4 La PAM afecta cerca de 5.9 por millón de habitantes. Esta entidad ha aumentado en las últimas dos décadas, lo cual podría explicarse por la posibilidad de realizar ANCA. El pico de ocurrencia está alrededor de 60 a 65 años y la relación hombre/mujer es de 1.6:1.5,6 La prevalencia en Colombia no es conocida, se han descrito alrededor de 21 casos casi todos con compromiso renal7-12, que es la presentación típica de la PAM. Sin embargo, también puede ser atípica con afectación pulmonar desencadenando el síndrome pulmonar-renal13, o debutando con manifestaciones dermatológicas14, oculares15, del sistema nervioso central o periférico16,17, y del sistema gastrointestinal18, entre otras.
Presentamos el caso de un adolescente con presentación atípica de PAM con compromiso renal, pulmonar, dermatológico y del sistema nervioso central.
Reporte de caso
Paciente masculino de 17 años de edad sin antecedentes epidemiológicos, que consulta por cuadro de disnea progresiva y tos con expectoración hemoptoica de 5 días de evolución. Dos meses antes de la enfermedad actual acudió al servicio de urgencias por tos productiva asociada con fiebre, astenia y adinamia. El cuadro se interpretó como neumonía bacteriana y fue manejado con antibióticos. Como antecedentes de importancia el paciente presentó 6 meses antes del ingreso glomerulonefritis membranosa que requirió diálisis peritoneal.
La radiografía de tórax del ingreso evidenció aumento de tamaño del corazón con opacidades nodulares finas mal definidas. La tomografía computarizada con cortes de alta resolución (TCAR) demostró nódulos centrilobulillares con densidad de vidrio esmerilado, densidades lineales ramificadas centrilobulillares de contornos mal definidos y áreas en vidrio esmerilado en “parches” (figura 1). En el diagnóstico diferencial se consideró infección por micobacterias y alteraciones parenquimatosas relacionadas con vasculitis de vaso pequeño.
En el curso de la enfermedad desarrolló lesiones vesicopapulares en la cara, correspondientes a vasculitis leucocitoclásica y necrosis fibrinoide en el estudio histológico (figura 2). La radiografía de tórax de control demostró progresión de la alteración parenquimatosa con consolidación multipolar y bilateral. En el lavado broncoalveolar se encontraron abundantes macrófagos con hemosiderina en su interior (90%), entremezclados con linfocitos (1%) y polimorfonucleares (8%).
Durante la hospitalización el paciente presentó deterioro clínico con hemorragia alveolar difusa y alteración del estado de conciencia. La TC de cráneo evidenció hemorragia subaracnoidea (cisterna perimesencefálica), con extensión intraventricular y ventriculomegalia secundaria.
El paciente falleció, se realizó autopsia clínica que evidenció encéfalo con circunvoluciones aplanadas, dilatación ventricular y hemorragia subaracnoidea asociada con vasculitis leucocitoclásica. Pulmones de consistencia firme, rojizos. En el estudio microscópico se observó intersticio engrosado con edema y capilaritis, alvéolos ocupados por sangre y hemosiderófagos los cuales resaltaron con la coloración de azul de Prusia (figura 3). Riñones de superficie irregular disminuidos de tamaño, en la histopatología glomerulonefritis con semilunas fibróticas, inflamación intersticial, atrofia tubular y necrosis fibrinoide. No se observaron granulomas. Se realizó perfil autoinmune con positividad para c-ANCA, el resto de estudios fueron negativos. El diagnóstico definitivo fue poliangeitis microscópica con afectación de riñón, piel, pulmón y sistema nervioso central, y como causa de muerte hemorragia subaracnoidea.
Discusión
La etiología de la PAM es incierta, se ha asociado con factores genéticos y medioambientales como infecciones, exposición a la sílice, al propiltiouracilo y a medicamentos.19 La fisiopatología de la enfermedad se caracteriza por la activación de moléculas de adhesión en los neutrófilos y la expresión de ANCA en la superficie. Estos neutrófilos después están en contacto con el endotelio generando degranulación de proteínas intracelulares, producción de citocinas y quimiocinas proinflamatorias, desencadenando especies activadas de oxígeno y producción de enzimas liticas. El sistema del complemento se activa por la vía alterna, generando activación de los neutrófilos a través del factor B y la properdina. Asimismo, la presencia de C5a está involucrada en la quimiotaxis de los neutrófilos20,21 que van a generar trampas extracelulares (NETosis) por un nuevo modelo de muerte celular. Estas proteínas se encuentran a nivel intracelular, lo cual aumenta la respuesta autoinmune, generando proteínas proinflamatorias y autoanticuerpos.5,19,22,23 El sistema inmune adquirido también juega un rol fundamental, los linfocitos B producen autoanticuerpos y los linfocitos T a través de la IL-23 activan la vía Th17 desencadenando daño tisular.20
La presencia de ANCA es fundamental para el diagnóstico, de hecho la presencia de MPO está presente en el 95% y de proteínas 3 en el 70% de los pacientes con PAM.24 Sin embargo, la presencia de ANCA también puede observase en otras vasculitis y enfermedades autoinmunes.19
Desde el punto de vista clínico afecta de manera principal al riñón, casi el 100%, en especial como una glomerulonefritis rápidamente progresiva. En algunos ocurre una forma atípica con nefritis tubulointersticial.25 Se manifiesta con proteinuria, hematuria microscópica y aumento de los niveles de creatinina y de la filtración glomerular.25
En el estudio histopatológico de la biopsia renal se puede observar necrosis de las asas capilares (capilaritis), proliferación extracapilar con formación de medias lunas, infiltrado intersticial y periglomerular (algunas veces simulando granulomas), arteritis necrosante y en la inmunofluorescencia se observa ausencia o escasez de inmunoglobulina y complemento en los glomérulos y los vasos renales, esta característica es denominada como “pauci-inmune”.25 La arteritis necrosante ocurre en menos del 20%3, mientras que la glomerulonefritis necrotizante y la capilaritis pulmonar necrotizante son más frecuentes. Las lesiones vasculares agudas tienen necrosis fibrinoide segmentaria con infiltración neutrofílica y leucocitoclásica. A los pocos días el infiltrado inflamatorio cambia de neutrófilos a monocitos, macrófagos y linfocitos. 26
Por otro lado, el compromiso pulmonar puede observarse entre el 25% al 90% de los pacientes con PAM.27 Cursan con tos, disnea, estridor, hemoptisis o dolor pleurítico, también se puede acompañar de síntomas como estenosis subglótica o traqueal, consolidación pulmonar, edema pulmonar, derrame pleural, pleuritis y consolidaciones. Pueden llegar a desarrollar fibrosis pulmonar e hipertensión arterial pulmonar.28 La hemorragia alveolar se considera el hallazgo más frecuente (12% a 50%).28 La mayoría de los pacientes presentan radiografía de tórax anormal, encontrando uno o más de los siguientes hallazgos: nódulos que se pueden cavitar, opacidades difusas, a veces con broncograma aéreo, atelectasia lobar o segmentaria, adenomegalias hiliares y opacidades pleurales. Los hallazgos más comunes son los nódulos que pueden ser únicos o múltiples con diámetro entre 1 y 10 cm, con una frecuencia de presentación de 20 a 90% de los casos de pacientes con poliangeítis microscópica con compromiso pulmonar. Otros hallazgos menos comunes son derrame pleural y neumotórax espontáneo. En la TAC de tórax encontramos múltiples nódulos pulmonares, por lo regular menos de 10, que miden desde milímetros hasta 10 cm y el 30 a 50% de estos se cavita, así como imagen en vidrio esmerilado u opacidades parcheadas o difusas debido a la hemorragia alveolar, patrón reticular, engrosamiento septal interlobular, panal de abejas, bronquiectasias, adenomegalias hiliares y derrame pleural.29
En la biopsia pulmonar se puede apreciar hemorragia alveolar con glóbulos rojos intraalveolares e intersticiales, capilaritis necrotizante alveolar hemorrágica y hemosiderosis intraalveolar.30 Al igual que en la inmunofluoresencia renal, a nivel pulmonar se observa ausencia o escasez de inmunoglobulina y complemento. La inflamación granulomatosa se observa sobre todo en la poliangeitis granulomatosa.29 En el lavado bronquioalveolar de los pacientes con hemorragia se observa líquido hemorrágico con frecuentes hemosiderófagos.31
El compromiso dermatológico secundario a PAM se ha reportado en el 20 a 70% y las lesiones más frecuentes son púrpura (78%), eritema (50%), pápulas (28%), livedo reticularis (17%), ampollas (11%), eritema urticarial (6%), hemorragias en astilla (6%) y úlceras (6%).14 A la histología se observa vasculitis leucocitoclásica, asociada con edema endotelial, extravasación de eritrocitos y degeneración fibrinoide de las paredes vasculares.14
El sistema nervioso puede verse afectado en 37% a 72% de los pacientes. Se ve más en el sistema periférico dados por la enfermedad o secundario al tratamiento inmunosupresor.29 Las alteraciones que se observan en el sistema nervioso central son infrecuentes, puede observarse hemorragia intracerebral o subaracnoidea, infartos pontino o cerebrales no hemorrágicos, o bien síndrome de leucoencefalopatía posterior reversible (SLPR).32,33 La hemorragia y la isquemia cerebral se desencadenan por el daño de las paredes vasculares y la posterior ruptura, principalmente en la poliangeítis granulomatosa y en el síndrome de Churg-Strauss.32
En nuestro paciente es interesante la presentación atípica, tanto por la ascendencia y edad de presentación, como el compromiso multisistemico con glomerulonefritis pauci-inmune, hemorragia pulmonar, vasculitis leucocitoclásica cutánea y hemorragia cerebral. Creemos que esta asociación no se ha informado previamente en la literatura médica. Los casos con compromiso de sistema nervioso central, renal y pulmonar se han publicado en pacientes asiáticos33–36, cursando con un mal pronóstico.
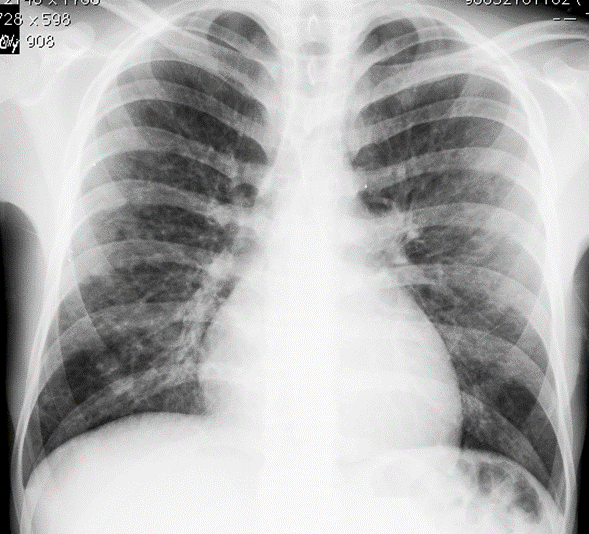
Figura 1A. TCAR corte axial. Nódulos centrilobulillares con densidad de vidrio esmerilado, de contornos mal definidos, multilobares, bilaterales.
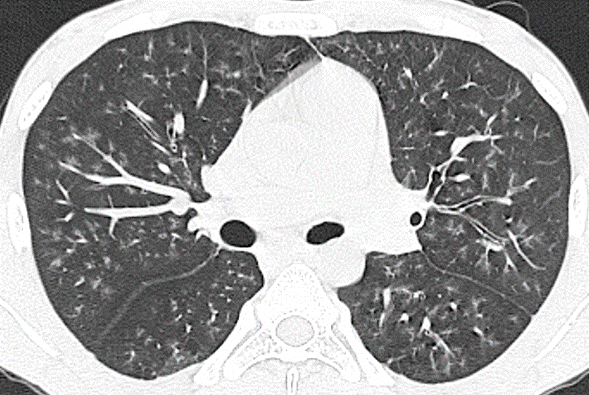
Figura 1B. Tórax con nódulos pequeños de contornos mal definidos, multilobares, bilaterales.
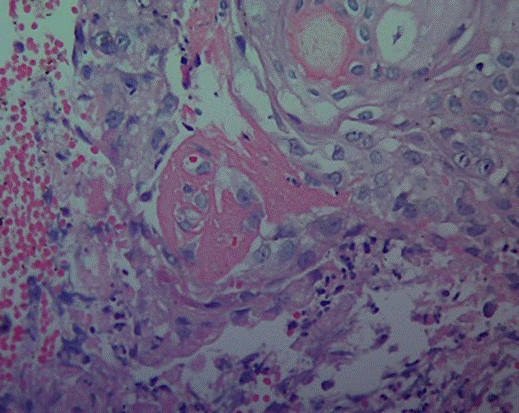
Figura 2. Piel: vasculitis leucocitoclásica y necrosis fibrinoide de los vasos. Hematoxilina-eosina 40x.
3
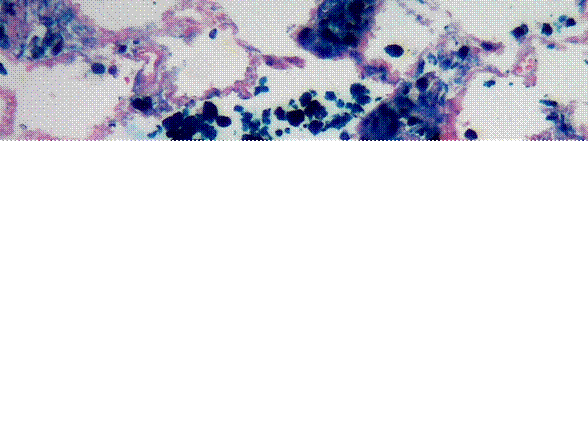
Figura 3. Pulmón: intersticio engrosado y hemosiderófagos intraalveolares. Azul de Prusia 20x.
Referencias
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
2. Falk RJ, Gross WL, Guillevin L, Hoffman GS, Jayne DRW, Jennette JC, et al. Granulomatosis with polyangiitis (Wegener’s): an alternative name for Wegener's granulomatosis. Arthritis Rheum. 2011;63(4):863–4.
3. Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology and prognosis. Q J Med. 1985;56(220):467–83.
4. Katsuyama T, Sada K-E, Makino H. Current Concept and Epidemiology of Systemic Vasculitides. Allergol Int. 2014 Oct 25;[Epub ahead of print].
5. Kallenberg CGM. The diagnosis and classification of microscopic polyangiitis. J Autoimmun. 2014;48-49:90–3.
6. Watts RA, Mooney J, Skinner J, Scott DGI, Macgregor AJ. The contrasting epidemiology of granulomatosis with polyangiitis (Wegener’s) and microscopic polyangiitis. Rheumatology (Oxford). 2012 May;51(5):926–31.
7. Álvarez F, Herrera FR, Contreras ÁS, D GMV, Cañas CA, Chalem M, et al. Vasculitis primarias : 62 años de historia en Colombia. Rev Colomb Reumatol. 2006;13(4):288–305.
8. Cantillo JDJ, Andrade RE. A propósito de dos nuevos casos en Colombia. Acta Med Colomb. 2008;33(2):84–9.
9. Camargo JP, Cantillo JDJ. Poliangeítis microscópica Trece años de su reconocimiento y 91 años a la sombra de la poliarteritis nodosa. Acta Med Colomb. 2008;33(2):90–100.
10. Argote AC, Merlano I. Poliangeítis microscópica Resumen Introducción Caso clínico. Rev Asoc Colomb Dermatol. 2013;21(2):175–9.
11. Jaramillo Garcia LF, Morales Naranjo SD. Poliangeitis microscópica, reporte de un caso. Rev colomb neumol. 2006;18(2):84–7.
12. Grajales CM, Cortés ÁA, Fernando L, Restrepo A, Escobar MR, Alonso L, et al. Poliangiítis microscópica : reporte de un caso con enfermedad renal , pulmonar y neurológica , y revisión de la literatura. Rev Colomb Reumatol. 2011;18:121–34.
13. Niles JL, Böttinger EP, Saurina GR, Kelly KJ, Pan G, Collins AB, et al. The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med. 1996;156(4):440–5.
14. Niiyama S, Amoh Y, Tomita M, Katsuoka K. Dermatological manifestations associated with microscopic polyangiitis. Rheumatol Int. 2008;28(6):593–5.
15. Hara A, Ohta S, Takata M, Saito K, Torisaki M, Ishida Y, et al. Microscopic polyangiitis with ocular manifestations as the initial presenting sign. Am J Med Sci. 2007;334(4):308–10.
16. Adams HP. Cerebral vasculitis. Handb Clin Neurol. 2014 Jan;119:475–94.
17. Vrancken AFJE, Said G. Vasculitic neuropathy. Handb Clin Neurol. 2013;115:463–83.
18. Latus J, Koetter I, Fritz P, Kimmel M, Biegger D, Ott G, et al. Gastrointestinal involvement in granulomatosis with polyangiitis and microscopic polyangiitis: histological features and outcome. Int J Rheum Dis. 2014;17(4):412–9.
19. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. Nature Publishing Group; 2014;10(8):463–73.
20. Chen M, Kallenberg CGM. ANCA-associated vasculitides--advances in pathogenesis and treatment. Nat Rev Rheumatol. 2010;6(11):653–64.
21. Kallenberg CGM, Heeringa P. Complement is crucial in the pathogenesis of ANCA-associated vasculitis. Kidney Int. Nature Publishing Group; 2013;83(1):16–8.
22. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15(6):623–5.
23. Matheus Merina LM, Parra-medina R, Ararat Sarria M, Díaz Galvis JA. Cell Death. Autoimmunity From Bench to Bedside. 2013. p. 219–34.
24. Terrier B, Saadoun D, Sène D, Ghillani P, Amoura Z, Deray G, et al. Antimyeloperoxidase antibodies are a useful marker of disease activity in antineutrophil cytoplasmic antibody-associated vasculitides. Ann Rheum Dis. 2009;68(10):1564–71.
25. Sinico RA, Di Toma L, Radice A. Renal involvement in anti-neutrophil cytoplasmic autoantibody associated vasculitis. Autoimmun Rev. 2013;12(4):477–82.
26. Jennette JC, Falk RJ, Heumatology EXR. The role of pathology in the diagnosis of systemic vasculitis. Clin Exp Rheumatol. 2007;25(1 Suppl 44):52–6.
27. Wilke L, Prince-Fiocco M, Fiocco GP. Microscopic polyangiitis: a large single-center series. J Clin Rheumatol. 2014;20(4):179–82.
28. Huang H, Wang YX, Jiang CG, Liu J, Li J, Xu K, et al. A retrospective study of microscopic polyangiitis patients presenting with pulmonary fibrosis in China. BMC Pulm Med. 2014;14:8.
29. Chung SA, Seo P. Microscopic polyangiitis. Rheum Dis Clin North Am. 2010;36(3):545–58.
30. Gómez-Puerta JA, Espinosa G, Morlà R, Cid MC, Cervera R. Interstitial lung disease as a presenting manifestation of microscopic polyangiitis successfully treated with mycophenolate mofetil. Clin Exp Rheumatol. 2009;27(1):166–7.
31. Collins CE, Quismorio FP. Pulmonary involvement in microscopic polyangiitis. Curr Opin Pulm Med. 2005;11(5):447–51.
32. Han S, Rehman HU, Jayaratne PS, Carty JE. Microscopic polyangiitis complicated by cerebral haemorrhage. Rheumatol Int. 2006;26(11):1057–60.
33. Tan J, Hussain A, Daiwajna R, Chai LK, Lim E, Han A. Microscopic polyangiitis complicated by intracerebral hemorrhage and pulmonary hemorrhage in a pediatric patient. Am J Case Rep. 2013;14:276–9.
34. Sasaki A, Hirato J, Nakazato Y, Tanaka T, Takeuchi H. [An autopsy case of P-ANCA-positive microscopic polyangiitis with multiple cerebral hemorrhagic infarction]. No To Shinkei. 1998;50(1):56–60.
35. Matsuda H, Harada T, Ando Y, Nakamori A, Nakatsuka S, Takama T, et al. [Case of microscopic polyangiitis accompanied by central nervous system symptoms and brain vasculitis observed histopathologically]. Nihon Jinzo Gakkai Shi. 2011;53(4):648–53.
36. Kimura H, Akutsu N, Shiomi R, Kohmura E. Subarachnoid hemorrhage caused by ruptured intracranial fusiform aneurysm associated with microscopic polyangiitis. Neurol Med Chir (Tokyo). 2012;52(7):495–8.