REPORTE DE CASO
ENCEFALITIS AUTOINMUNE: EXPERIENCIA DE UNA SERIE DE CASOS
Fecha recibido: enero 23 de 2018
Fecha aceptado: noviembre 8 de 2018
Nathalie Mariño MDa, Javier Triana MDb, VíctorNieto MDc, Jean Paul Vergara MDb, Miguel Silva MDb, Eduardo Palacios MDb,d
Correo electrónico autora principal Dra. Nathalie Mariño: jmarinoenriquez@gmail.com
a Neurología, Fundación Universitaria de Ciencias de la Salud. Bogotá DC, Colombia.
b Servicio de Neurología, Hospital de San José. Bogotá DC, Colombia.
c Unidad de Cuidado Intensivo Adulto, Hospital de San José. Bogotá DC, Colombia.
d Miembro de la Sociedad de Cirugía de Bogotá, Fundación Universitaria de Ciencias de la Salud, Bogotá DC, Colombia.
Resumen
La encefalitis límbica autoinmune se define como un desorden inflamatorio severo en el cerebro que genera una encefalopatía rápidamente progresiva por lo regular en menos de 6 semanas. Se han descrito anticuerpos contra epítopes de proteínas sinápticas de superficie o contra proteínas intracelulares. Se reportan seis casos de encefalitis autoinmune con la descripción de cada uno incluyendo las manifestaciones típicas, alteraciones imagenológicas, curso clínico, complicaciones durante la hospitalización y respuesta al tratamiento. En dos de los casos se logró confirmar mediante el aislamiento de los anticuerpos. Uno falleció durante su hospitalización. Nuestra experiencia muestra un alto riesgo de secuelas y refractariedad al tratamiento que dependen del tiempo de inicio de la terapia.
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS. Este es un artículo Open Access
bajo la licencia CC BY-NC-ND (http://creativecommons.org/licenses/by-nc-nd/4.0/).
AUTOIMMUNE ENCEPHALITIS: A CASE SERIES
ABSTRACT
Autoimmune limbic encephalitis is defined as a severe inflammatory neurological disease which causes a rapidly progressive encephalopathy, usually in less than six weeks. An association with autoantibodies to extracellular epitopes or antibodies to intracellular synaptic proteins has been described. Six cases of autoimmune encephalitis are here presented, including typical findings, imaging alterations, clinical features, inpatient stay complications and response to treatment. Two cases were confirmed by autoantibody testing. One patient died during hospitalization. Our experience show, high risk of sequelae and unresponsive states to treatment which depended on how soon the treatment was initiated.
Key Words: autoimmune encephalitis; atypical behavioral symptoms; epilepsy
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS. This is an open access article under the CCBY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Introducción
La variedad autoinmune es una causa de encefalitis recientemente descrita, generando alteraciones neuropsiquiátricas a menudo seguidas de deterioro progresivo del estado de conciencia, epilepsia refractaria y coma.1 Dentro de los síndromes encefalíticos se distinguen dos grandes categorías: la encefalitis autoinmune paraneoplásica asociada con autoanticuerpos onconeurales contra antígenos neuronales intracelulares y la autoinmune asociada con autoanticuerpos contra antígenos de superficie o sinápticos como los anti NMDA, los cuales fueron reportados por primera vez en 2007 y en el momento son reconocidos como una de las causas más comunes de dicha afección.2-4 En la clínica estos desórdenes son un reto para el personal médico debido a la variedad de manifestaciones neurológicas5 y la pobre especificidad de las características del LCR, del monitoreo electroencefalográfico y las neuroimágenes, así como la dificultad para lograr el procesamiento de los anticuerpos contra antígenos específicos ya que no se encuentran disponibles en la mayoría de laboratorios a nivel nacional, aunque es importante aclarar que estos anticuerpos a pesar de ser específicos no están incluidos en los criterios diagnósticos para encefalitis autoinmune6, probablemente por las limitaciones de las instituciones para detectarlos.
La morbilidad y mortalidad secundaria a esta patología y los altos costos derivados de hospitalizaciones prolongadas, requerimiento de unidades de cuidado intensivo y necesidad de terapias inmunomoduladoras costosas7, ha incrementado el interés del personal médico en cuanto a su diagnóstico y manejo, requiriendo descartar causas no infecciosas de encefalitis que se han reportado en cerca de 50%.8 Una de las características de este tipo de encefalitis que ha sido mencionada en algunas publicaciones es la buena respuesta a la inmunoterapia9, sin embargo esta no es la norma y se reportan casos refractarios.6
En esta publicación tenemos la oportunidad de reportar seis casos probables de encefalitis autoinmune que se han diagnosticado en el Hospital de San José de Bogotá, entre los años 2014 a 2016, dos de ellos con confirmación de anticuerpos anti NMDA y anti VGKC (anti LGI1). En todos los casos se excluyeron causas paraneoplásicas con los recursos que se tienen en nuestra institución. El ingreso a UCI de estos pacientes se debió a alteraciones severas del sensorio, necesidad de protección de la vía aérea y estado epiléptico como se ha reportado en la literatura10, así mismo se trasladaron para inicio y vigilancia de la terapia con plasmaféresis.
Objetivos
Describir las manifestaciones clínicas, las anormalidades imagenológicas, el procesamiento de anticuerpos, respuesta al tratamiento instaurado y secuelas neurológicas de los casos de encefalitis autoinmune presentados en el Hospital de San José de Bogotá entre los años 2014 y 2016.
Metodología
Estudio de serie de casos. Se incluyeron todos los pacientes mayores de 18 años con sospecha clínica durante su hospitalización de encefalitis autoinmune en el Hospital de San José de Bogotá durante 2014 a 2016.
Resultados
CASO 1
En octubre 2014, ingresa paciente femenina de 70 años, ama de casa, por cuadro de un día de evolución consistente en alteraciones neuropsiquiátricas con cambios en el comportamiento, agresividad, agitación psicomotora de causa no tóxica, infecciosa ni metabólica. Como antecedentes personales se resalta hipertensión arterial, tabaquismo y desde hace 8 meses fallas en la memoria de corto plazo no estudiada. Al ingreso a nuestro servicio presenta desorientación, alteraciones en el lenguaje, ideas delirantes de persecución, pensamiento perseverante, juicio y raciocinio comprometido y fallas de memoria de corto y largo plazo; adicionalmente presencia de reflejos patológicos glabelar y prensión palmar. Se realizaron neuroimágenes sin evidencia de hallazgos patológicos y estudio de líquido cefalorraquídeo sin alteraciones, una videotelemetría con lentificación de ritmos de fondo con puntas agudas pseudoperiódicas de predominio frontal izquierdo. La paciente persistió con alteraciones neuropsiquiátricas sin documentarse causa, habiendo descartado neoplasia. Se consideró que podría estar cursando con un cuadro de origen autoinmune y se decidió a los 7 días de ingreso iniciar pulsos de metilprednisolona intravenosa sin respuesta a este manejo. Después cursó con deterioro del estado neurológico, persistieron las alteraciones comportamentales asociadas con compromiso severo del ciclo sueño vigilia y síntomas parkinsonianos. Se decidió a los doce días del ingreso, trasladarla a la unidad de cuidados intensivos para iniciar terapia con plasmaféresis, realizando 5 sesiones con mejoría de la alteración del patrón del sueño pero con persistencia del parkinsonismo y las alteraciones neuropsiquiátricas. Debido a ello 7 días después de terminar primer ciclo de plasmaféresis, se consideró otro con 5 sesiones adicionales, sin embargo no se logró respuesta clínica. Se solicitó proteína 14-33, anticuerpos anti NMDA y VGKC los cuales no fueron autorizados por su aseguradora de salud.
Durante la hospitalización presentó infección de vías urinarias, sangrado digestivo bajo e inestabilidad hemodinámica, disautonomías, trastorno electrolítico severo, picos febriles asociados y finalmente fallece a los 3 meses de hospitalización.
CASO 2
Paciente de 28 años que ingresa en enero de 2014 por cuadro clínico de 8 días de evolución de astenia y adinamia, hiporexia, sensación de malestar general asociado con episodios eméticos y luego con alteración aguda del estado de conciencia dado por somnolencia y presencia de crisis tónicas con movimientos clónicos, posterior a esto presentó agitación psicomotora y agresividad. No se encontraron antecedentes de importancia, inicialmente se manejó como meningoencefalitis y recibió aciclovir y ceftriaxona. En el líquido cefalorraquídeo se encontró una leve proteinorraquia sin otras alteraciones (proteínas 71 mg/dL, recuento celular leucocitos 5 x mm3, hematíes 150 x mm3, glucosa 112 mg/dL, serología no reactiva, tinta china negativa, índice de glucosa: 0.5). Se solicitaron estudios de extensión en LCR, PCR para herpes tipo I y II, Epstein barr, TBC, látex para criptococo, anticuerpos antitiroideos y perfil autoinmune los cuales se consideraron negativos. Se trasladó a la unidad de cuidados intensivos debido al mayor deterioro del estado de conciencia con persistencia de crisis epilépticas de inicio focal, con dos estudios electroencefalográficos que mostraron actividades lenta difusa y paroxística multifocal más evidentes sobre el hemisferio derecho. La resonancia de cerebro, simple y contrastada, mostró hiperintensidades en T2 y FLAIR en caudado, putamen, temporal e insular bilaterales (figura 1). Se consideró estado epiléptico refractario secundario a una encefalitis autoinmune y se solicitó autorización para investigar anticuerpos antiNMDA Y VGKC. Se inicia manejo con pulsos de metilprednisolona intravenosa, con segunda punción lumbar sin alteraciones. Al tercer día de manejo con metilprednisolona hubo mejoría del estado de conciencia, aunque pronto aparecieron de nuevo fluctuaciones, alodinia marcada en miembro inferior izquierdo, prurito severo en genitales, conductas desinhibidas, inatención, confusión, movimientos erráticos de las 4 extremidades con posturas distónicas en miembros inferiores y empeoramiento de la sensación de prurito generalizado y en genitales. La paciente se trasladó a la unidad de cuidados intermedios con mejoría de patrón del comportamiento, se definió realizar estudios para descartar patología neoplásica y se continuó manejo con prednisona 60 mg/día. Posteriormente presento mejoría del comportamiento y del estado de conciencia y se controlaron las crisis convulsivas. Durante la hospitalización no fue autorizada por su aseguradora la toma de anticuerpos antiNMDA y VGKC.
En los controles ambulatorios persistió con crisis focales sensitivas del lóbulo temporal, recibió manejo con antiepilépticos y continuó en un estado encefalopático de leve a moderado. Se realizó control de perfil autoinmune a los 6 meses encontrando resultados paraclínicos sugestivos de lupus eritematoso sistémico.
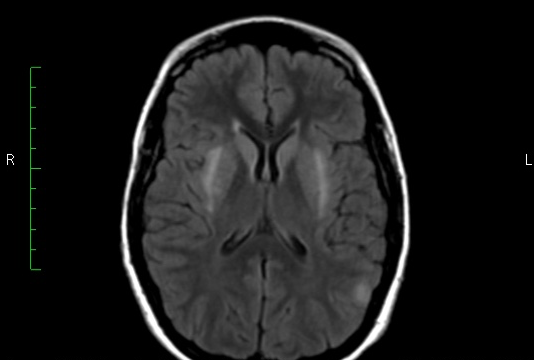
Figura 1. Secuencia T2-FLAIR mostrando hiperintensidad en el ganglio basal bilateral con mayor compromiso de putamen.
CASO 3
Paciente de 66 años de edad, ocupación abogado, quien acude en mayo 2014 al servicio de consulta externa con cuadro clínico de 5 meses de evolución consistente en deterioro cognitivo, somnolencia diurna, disminución de la capacidad intelectual, abandono del trabajo, asociado con 2 crisis convulsivas. Como antecedente el paciente había presentado episodios de hiponatremia euvolémica desde hace 5 meses de origen desconocido. Ingresó con minimental test 20/30 y sodio sérico de 129 mmol/L. Se decidió hospitalizar para estudio. Durante su estancia presentó aumento de la frecuencia ictal hasta 20 crisis al día y se documentó una secreción inadecuada de hormona antidiurética la cual resolvió. Las resonancias magnéticas cerebral simple y contrastada no mostraron alteraciones y la punción lumbar reportó un LCR sin alteraciones. Se solicitaron proteína 14-33, anticuerpos anti NMDA y VKGC. Se consideró la encefalitis autoinmune por anticuerpos anticanales de potasio por su asociación con hiponatremia y se iniciaron estudios de marcadores neoplásicos, perfil autoinmune y búsqueda de neoplasia, los cuales se reportaron finalmente negativos. Se decidió el manejo inicial con pulsos de esteroides con respuesta parcial y posterior a esto se inició plasmaferesis en la unidad de cuidados intensivos, en donde se realizaron 6 sesiones. Hubo mejoría en la frecuencia de crisis convulsivas y en el ciclo sueño vigilia, persistiendo con alteraciones cognitivas. Se continuó el manejo con prednisona 1 mg/k/día y no se decidió escalar a otra terapia inmunomoduladora. Al egreso presentaba encefalopatía leve, deterioro cognitivo moderado y mioclonias ocasionales.
La paciente regresa a control por consulta externa sin autorización aun de anticuerpos solicitados, los cuales se solicitan de nuevo. La proteína 14-33 fue negativa.
CASO 4
Paciente de 51 años de edad que ingresó en enero 2014 con cuadro clínico agudo de un día de evolución consistente en mutismo, apatía, sensación de angustia, llanto y mioclonias. Se realizó RMN cerebral simple y contrastada en la cual no se evidenciaron alteraciones, LCR sin anormalidades (leucocitos 0, hematíes 3, proteínas 176 mg/dL, glucosa 49 mg/dL, glicemia 88 mg/dL, índice de glucosa 0.55). Inicialmente recibió manejo con aciclovir considerándose encefalitis de etiología viral y se solicitaron pruebas de extensión PCR para herpes virus tipos I y II, Epstein barr los cuales finalmente fueron reportados negativos. Persistió con periodos de agitación psicomotora a pesar del manejo con aciclovir. Se realizó videotelemetría de 24 horas la cual presentó 10 episodios que inician con movimientos mioclónicos de miembro superior derecho y luego bilateral, sin síntomas disautonómicos, conservando siempre la apertura ocular y luego actividad focal de predominio frontocentral izquierdo que evolucionaba en frecuencia y amplitud hasta generalizar en polipuntas con un máximo bifrontal. Se consideró una probable encefalitis autoinmune y se iniciaron pulsos de metilprednisolona intravenosa a los 4 días de ingreso a la institución. Se realizaron estudios de extensión para descartar etiología paraneoplásica, los cuales fueron negativos en ese momento.
Durante el manejo con pulsos de metilprednisolona presentó buena respuesta a esta terapia, con mejor interacción con el examinador y resolución completa del estado de agitación psicomotora. Por la mejoría clínica fue dada de alta, con solicitud de anticuerpos en LCR en forma ambulatoria. En el último control aun no se han autorizado anticuerpos, con secuelas neurológicas dadas por limitación para la comunicación con el entorno, depresión y encefalopatía leve.
CASO 5
Paciente femenina de 47 años de edad, abogada, previamente sana, quien ingresa en marzo 2016 por cuadro neuropsiquiátrico agudo de 5 días de evolución consistente en mutismo, risas inmotivadas, estereotipias oromandibulares y manuales, cambios conductuales y disfunción ejecutiva. Se documentó además hipersomnia 3 meses antes del ingreso. Al examen físico se evidenció postura flácida con cabeza en versión derecha sin respuesta al llamado y con respuesta extensora izquierda. Se realizó punción lumbar con reporte de pleocitosis linfocitaria (leucocitos 72 xmm3, glucosa 51 mg/dL, proteínas 46 mg/dL, neutrófilos 2%, linfocitos 98%, hematíes 4, glucosa 89 mgdL, serología no reactiva). Se inició manejo con aciclovir, al día siguiente presentó postura tónica de miembro superior izquierdo y versión cefálica a la izquierda con nistagmus de fase rápida a la derecha, con RMN de cerebro, simple y contrastada, con evidencia de múltiples lesiones hipointensas en T1 e hiperintensas en T2 y FLAIR a nivel de ganglios basales, temporal izquierdo y mesencefálico (figura 2). La paciente continuó con alteraciones neuropsiquiátricas a pesar de manejo con aciclovir, por lo cual dada la evolución y las neuroimágenes se definió como encefalitis de probable origen autoinmune, se solicitaron anticuerpos antiNMDA, VKGC y proteína 14-33. Se iniciaron pulsos de metilprednisolona (5 días) sin lograr repuesta y con deterioro neurológico progresivo, la videotelemetría con hallazgos anormales severos por presencia de ondas de actividad lenta de fondo, ondas trifásicas, actividad paroxística generalizada, seguida de disminución del voltaje, sugestivo de un proceso encefalopático severo y actividad epileptiforme frecuente. Requirió soporte en la unidad de cuidados intensivos, en donde se descartó patología neoplásica. A los 8 días del ingreso se inició manejo con plasmaféresis completando 5 sesiones sin mejoría significativa, 8 días después debido a la persistencia de requerimiento de soporte ventilatorio y nutricional se realiza traqueostomía y gastrostomía.
Las resonancias de cerebro simple y contrastada de control revelaron disminución de las lesiones pero con persistencia del severo compromiso neurológico. Se recibe reporte de VKGC y proteína 14-33 negativos, pero antiNMDA positivos. Se plantea manejo con ciclofosfamida, pero no se inició el ciclo de inducción por múltiples procesos infecciosos no resueltos entre ellos neumonía y traqueítis. Se definió después como no candidata a otra medida y se continua manejo con inmunomodulador oral, azatioprina y prednisona oral. A los dos meses de hospitalización se da salida con programa de plan hospitalario domiciliario.
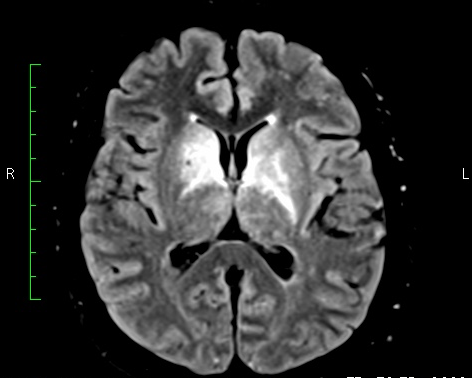
Figura 2. Secuencia T2-FLAIR mostrando hiperintensidades a nivel de ganglios de la base en forma bilateral.
CASO 6
Paciente de24 años, que ingresa en junio 2016 por cuadro clínico de 6 meses de evolución consistente en alteración del patrón del sueño dado por insomnio, cambios comportamentales, alteración de la memoria de corto plazo, asociados con crisis distónicas braquiofaciales y generalizadas. El paciente fue estudiado antes en otro hospital con sospecha diagnóstica de encefalitis autoinmune, en donde se manejó con pulsos de metilprednisolona intravenosa 1 gramo día por 5 días sin respuesta satisfactoria. Acude a esta institución por persistencia de la sintomatología, aumento de las alteraciones cognitivas y de la frecuencia de crisis distónicas braquiofaciales. Al ingreso paciente bradipsíquico con lenguaje hipofluente, con alteración severa de la memoria de corto plazo y crisis distónicas braquiofaciales cada minuto. Se realizó RMN cerebral simple y contrastada sin alteraciones, reporte de LCR normal. Se decidió traslado a la unidad de cuidados intensivos para iniciar el manejo con plasmaféresis, 5 sesiones, sin complicaciones durante el procedimiento y con mejoría en la evaluación cognitiva. Sin embargo, posterior a la finalización de la terapia la paciente presenta mayor compromiso de la memoria y la fluencia, con funciones ejecutivas relativamente conservadas y persistencia de crisis distónicas braquiofaciales asociadas con disautonomías, hay alteraciones de la frecuencia cardiaca y de la saturación de oxígeno, sugiriendo mayor compromiso temporal mesial que frontal. Se realiza una nueva RMN, cerebro simple y contrastado, evidenciando zona hiperintensa en secuencias T2 y FLAIR en el lóbulo medio temporal bilateral(figura 3). Se realizó estudio electroencefalográfico de 6 horas sin evidencia de crisis pero con compromiso de sueño logrando solo estadio I. Debido a la poca respuesta a la terapia de plasmaféresis, a los 10 días se decide iniciar fase de inducción con ciclofosfamida con el primer bolo de un gramo IV, el cual se repetiría cada mes para un total de 6 bolos. Posterior a esta terapia hay mejoría en la frecuencia de crisis y la alteración cognitiva por lo cual se define alta médica. Se solicitaron anticuerpos antiNMDA y VKGC en forma ambulatoria, con reporte de anticuerpos VKGC positivos durante su control por consulta externa. Se ordena segundo bolo de ciclofosfamida al mes. La paciente reingresa a los dos meses por aumento de la frecuencia de las crisis con alteración de la calidad de vida. Se decidió realizar nuevo ciclo de plasmaféresis en la unidad de cuidados intensivos, con un total de 10 sesiones sin mejoría clínica, por lo cual se procedió con rituximab con una segunda administración a los 15 días. Hubo mejoría en los primeros días posterior a la administración del medicamento, pero reinicia a los pocos días con distonia braquiofacial y deterioro cognitivo. Así mismo se solicitó PET SCAN corporal por refractariedad a los diferentes manejos, el cual aún se encuentra en espera de autorización. Continuamos manejo por el momento con prednisolona a 1 mg/k/día más azatioprina 50 mg cada 12 horas.
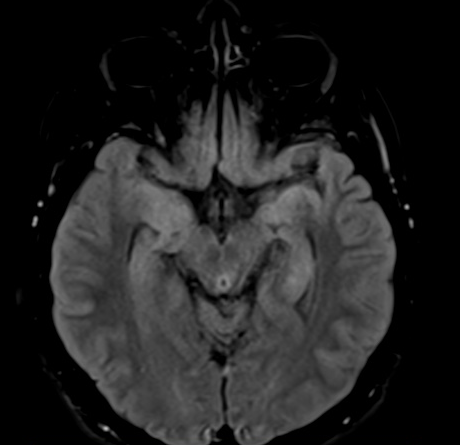
Figura 3. Secuencia T2-FLAIR mostrando hiperintensidad bilateral a nivel de lóbulos medios temporales.
Discusión
La encefalitis límbica autoinmune ha sido definida como un desorden inflamatorio severo que genera una encefalopatía rápidamente progresiva (usualmente en menos de 6 semanas)6 y la cual incluye un heterogéneo grupo de síndromes
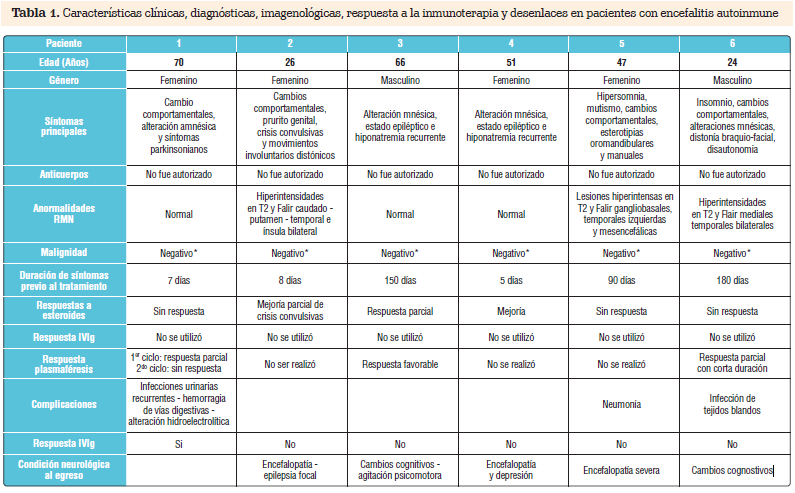
encefalíticos, con dos grandes categorías: la encefalitis autoinmune paraneoplásica asociada con los autoanticuerpos onconeurales contra antígenos neuronales intracelulares por ejemplo Hu, Ma2, CRMP-5.11-13 y que están asociados especialmente con cáncer de pulmón y tumores testiculares14; y segundo la encefalitis autoinmune asociada con autoanticuerpos contra antígenos de superficie o sinápticos incluyendo anticomplejo canales de potasio voltaje dependientes VGKC tales como el LGI1y antirreceptores Caspr2, NMDA, AMPA, GABA, contra canales de calcio voltaje dependientes VGCC, entre otros.3,4,15,16 La morbilidad y mortalidad secundaria a esta patología y los importantes costos que implican estadías prolongadas de hospitalización, requerimiento de unidad de cuidados intensivos y necesidad de terapias inmunomoduladoras costosas, ha incrementado el interés del personal médico en estudiar las causas no infecciosas de encefalitis, las cuales según el proyecto de encefalitis de California indicó que corresponden al 50% de los casos.8 En esta oportunidad presentamos el reporte de una serie de casos con este diagnóstico clínico en dos años de estudio. En la tabla 1, se mencionan las características principales de sus manifestaciones clínicas, las anormalidades imagenológicas, el procesamiento de anticuerpos, la respuesta al tratamiento instaurado y las secuelas neurológicas.
Las condiciones autoinmunes no son una causa común de encefalitis17, pero cuando se presentan suelen ser catastróficas. El reconocimiento de los síndromes clínicos, la disponibilidad de los métodos diagnósticos y el inicio temprano de la inmunoterapia pueden llevar a resultados favorables en este síndrome.2,7,15 Existen
criterios diagnósticos para la EA que son exclusivamente dependientes de las pruebas de anticuerpos y la respuesta a la inmunoterapia18, sin embargo este concepto no es realista debido a que las pruebas de anticuerpos son poco accesibles y en muchas instituciones como la nuestra, la toma de estas pueden tardar semanas tanto por la autorización por parte de la aseguradora, como en el reporte del mismo. Así mismo, la negatividad de estas pruebas no excluye la posibilidad de que el proceso sea inmunomediado.6 En cuanto a la respuesta de la terapia inmunomoduladora también se ha mencionado que la respuesta favorable no es la norma y en algunos casos es necesario practicar terapias prolongadas e intensivas para lograr una respuesta significativa.9 Muchas instituciones no cuentan con estos tratamientos y tienen poca experiencia en la implementación de terapias de segunda línea, por lo cual este criterio no está ampliamente aceptado en la actualidad.
En una reciente revisión de Graus y col.6 se hace énfasis en estas limitaciones, proponiendo que puede formularse un posible diagnóstico cuando se cumplen los tres criterios siguientes: 1). inicio subagudo (rápida progresión menor de 3 meses de déficit en la memoria de trabajo, alteraciones del estado mental o síntomas psiquiátricos); 2). al menos uno de estos factores: hallazgos nuevos focales en el SNC, convulsiones no explicadas por una causa conocida previa, pleocitosis en el estudio de LCR (más de 5 células) y cambios en RMN sugestivos de encefalitis, y 3) una exclusión exhaustiva de causas alternativas, lo cual deja por fuera la respuesta inmunomoduladora y la necesidad de toma de anticuerpos. Por otro lado en un reporte de casos de la Clínica Mayo10 para la selección de pacientes con diagnóstico de encefalitis autoinmune, se tuvo en cuenta tanto la presencia de al menos un anticuerpo neuronal clásico o si estos estuviesen negativos la presencia de más de 3 de los siguientes hallazgos: fenotipo clínico o radiológico clásico de encefalitis límbica , LCR inflamatorio (proteínas > 70 mgdL, > 5 células blancas, > 4 bandas oligoclonales, elevada síntesis de IgG y alto índice de IgG), más de una enfermedad autoinmune coexistente, diagnóstico de cáncer contemporáneo (causa paraneoplásica), mejoría neurológica atribuida a la inmunoterapia y neuropatología consistente con causa paraneoplásica o autoinmune, lo cual representa alternativas diagnósticas en la definición de estos pacientes. En nuestro estudio en menos de 30% (2 de 6 pacientes) fue posible realizar los anticuerpos en LCR y conocer el resultado, mientras que en los demás la prueba fue solicitada para anticuerpos contra antígenos de superficie, pero sin autorización por parte de la aseguradora de salud de cada paciente, lo que implica que el diagnóstico se basó en las características clínicas, las neuroimágenes, la respuesta a terapia inmunomoduladora y la exclusión de otras patologías.
La EA puede manifestarse con distintos síndromes que complican su reconocimiento, sin embargo la clásica presentación consiste en un cuadro subagudo (días a pocas semanas) de disminución progresiva del nivel de conciencia1,10 a menudo con fluctuaciones y alteraciones cognitivas, en especial déficit de las memorias de corto plazo y de trabajo6, alteraciones en el comportamiento, psicosis, convulsiones, disquinesias sobre todo orofaciales, distonías faciobraquiales, rigidez, signos extra piramidales y disfunción autonómica.7,17 En nuestro grupo de pacientes los cambios comportamentales y las alteraciones amnésicas fueron las más frecuentes, dos ingresaron por mutismo y también manifestaron trastornos del patrón del sueño por hipersomnia e insomnio, también reportados en la literatura.6
Últimamente se han tratado de asociar las diferentes presentaciones clínicas con cada anticuerpo identificando espectros específicos.4 El caso 5 en quien se encontraron anticuerpos antiNMDA en LCR, cumple con ciertas características como el género, ya que estos anticuerpos son más frecuentes en mujeres, lo cual se ha informado en el reporte de casos más grande, en el cual el síndrome antiNMDA se presentó en 91 mujeres y 9 hombres.19,20 Aunque se realizaron estudios para descartar neoplasia no se encontró evidencia de asociación con teratoma ovárico, haciendo parte de ese 40% de pacientes que no tienen relación con tumores.19,21 Otras malignidades asociadas con estos anticuerpos incluyen teratomas testiculares y mediastinales, linfoma de Hodgkin, carcinoma de células pequeñas y neuroblastoma.22,23 En las últimas semanas la paciente se encuentra en espera de realización de PET SCAN.
Las manifestaciones clínicas en pacientes con estos anticuerpos se generan por una reducción en la liberación del GABA por la neurona presináptica, que después se traduce en una liberación de glutamato, dopamina y la desregulación que conduce a excitotoxicidad clásica de este tipo de encefalitis24, en la cual se ha reportado un pródromo (visto en nuestro caso probable número 2) consistente en cefalea, fiebre y enfermedad viral no especifica 2 semanas antes de la presentación, frecuentes síntomas psiquiátricos19 como psicosis a menudo con ansiedad, paranoia, alteraciones del sueño (caso 5 con hipersomnia), delusiones y manías y alteraciones en el lenguaje variando entre ecolalia y mutismo17(caso 5). Otras manifestaciones descritas incluyen movimientos hiperquinéticos estereotipados, disquinesias orofaciales y linguales, así como trastornos del movimiento hiperquinéticos incluyendo desviación oculógira, opistótonos, corea, distonías y opsoclonus mioclonus. 15,25,26 En nuestro caso confirmado se evidenciaron estereotipias oromandibulares y manuales durante la hospitalización. Los casos 1,2 y 4 cuyo género es femenino también comparten características clínicas que nos hicieron pensar en un síndrome dado por anticuerpos antiNMDA, el caso 1 por ejemplo tiene a favor de encefalitis autoinmune un rápido deterioro neurológico de un día de evolución, lo cual más que una enfermedad por priones sugiere un proceso inflamatorio agudo. Se ha documentado además importante inestabilidad autonómica causando alteraciones respiratoria y hemodinámica como ocurrió en los casos 1 y 5. En raras oportunidades se puede presentar bradicardia que ha requerido marcapasos y necesidad de ventilación mecánica por hipoventilación central.27 Los anticuerpos antiNMDA pueden detectarse en sangre o en LCR y los niveles pueden ser monitoreados para definir en ciertos casos una respuesta pobre o satisfactoria al tratamiento13, sin embargo en nuestra institución no se ha podido realizar este control.
En los casos 1,2 y 4 con sospecha de encefalitis por anticuerpos antiNMDA vemos cómo los resultados son variables en cada una de ellas, el primero fallece, el segundo caso presenta un proceso encefalopático moderado y el 5 con confirmación de anticuerpos presenta un curso prolongado que requirió traqueostomía y gastrostomía, con infecciones recurrentes y una severa encefalopatía, lo que se relaciona con algunas publicaciones que informan recuperación incompleta en 45% de pacientes, pobre en 30% y una tasa de mortalidad de 4%.15,21,28 El cuarto caso logró una buena respuesta con terapia de primera línea mediante pulsos de esteroides siendo el único que se relaciona con la buena respuesta a la terapia y secuelas leves mencionados por algunos estudios como característica de estos anticuerpos.19 Un estudio multicéntrico observacional demostró que solo 53% respondió a terapia con esteroides, IVIG o terapia con plasmaferesis, con una frecuencia de recaídas entre 12 y 24%,9 encontrando como factores relacionados con mal pronóstico el inicio tardío de la terapia inmunomoduladora y ausencia de diagnóstico de tumor relacionado, ya que este podría ser sometido a resección quirúrgica con mejoría de los síntomas.29
En el caso 6 encontramos la positividad para anticuerpos VGKC, aunque hay evidencia reciente que sugiere la existencia de otros receptores como el LGI130 y el autoantígeno CASPR2 (contactin-associated protein-like 2) asociado con epilepsia refractaria, encefalitis y disfunción nerviosa periférica, o bien la combinación de los dos conocida como síndrome de Morvan.17,31,32 En la actualidad la solicitud de este anticuerpo en nuestra institución sigue siendo con la terminología antigua VGKC. El reporte del laboratorio de este paciente se conoció a los dos meses de solicitado, cuando asistió a consulta ambulatoria, pero no se informaron los niveles séricos, que suelen ser altos (>1000 pM) en condiciones patológicas del SNC comparados con las cifras bajas cuando se trata de hiperexcitabilidad del sistema periférico.33 Recordando este caso 6 con confirmación de anticuerpos, es un paciente joven de 23 años que presenta alteraciones del patrón del sueño y después del comportamiento, de la memoria a corto plazo y con crisis distónicas braquiofaciales, controversialmente llamadas convulsiones debido a que no se encuentran cambios electroencefalógraficos asociados34, por lo cual pueden llamarse como eventos motores. Presentó además alteración del sueño REM, inestabilidad autonómica y cambios en la RMN cerebral a nivel temporal medial, muy bien descritos en la literatura.15,35-37 Algunos pacientes con anticuerpos VGKC han sido reportados con malignidades asociadas, incluyendo timomas y cáncer de pulmón, aunque típicamente se considera una entidad paraneoplásica.38 En nuestro caso la mala respuesta a los esteroides, a las múltiples sesiones de plasmaféresis en UCI y la terapia con ciclofosfamida, nos llevó a considerar la solicitud de PET SCAN corporal para definir un tumor atípico que estuviera generando la refractariedad al manejo así como las recaídas; además la terapia más agresiva con rituximab administrada en el último mes con franca mejoría en los primeros días terminó a las pocas semanas con la reaparición de los eventos motores y el estado cognitivo.
El caso número 3 a pesar de tener los resultados de los anticuerpos específicos, su cuadro clínico consistente en alteraciones mnésicas, epilepsia refractaria e hiponatremia recurrente fue tratado como encefalitis autoinmune por anticuerpos VGKC, con una respuesta parcial a pulsos de esteroides y una muy favorable con la plasmaféresis.
En algunos casos se ha visto la coexistencia de anticuerpos antiNMDA y antiVGKC en pacientes que han requerido una estadía prolongada en UCI.10 Otros anticuerpos reportados en la literatura son los antiGABAR que causan encefalitis límbica con disfunción en la memoria y convulsiones prominentes39, anticuerpos antiAMPAR con encefalitis límbica caracterizada por psicosis atípica y relacionada en un 70% con el tumor pulmonar de células pulmonares pequeñas.15 Otros anticuerpos relacionados con encefalitis límbica paraneoplásica como antiHU y antiMA232 no se han podido investigar en nuestra institución.
Los cambios imagenológicos se han descrito para cada tipo de anticuerpos40, en el caso de encefalitis autoinmune por anticuerpos antiNMDA se han reportado anormalidades solo en 23 a 50%9,40, principalmente con compromiso de los lóbulos frontal, parietal y medial temporal, así como del giro del cíngulo, tálamo, cerebelo, tronco encefálico y ganglios basales.41-43 Estos hallazgos fueron encontrados en la resonancia cerebral del caso 5 con evidencia de múltiples lesiones hipointensas en T1 e hiperintensas en T2 y FLAIR a nivel de ganglios basales, lóbulo temporal izquierdo y mesencéfalo. También se han reportado cambios en el PET con hipometabolismo de la cabeza del caudado y núcleo lenticular, lo cual no se ha podido evidenciar en ninguno de nuestros pacientes. En el caso número 6 positivo para anticuerpos antiVGKC se observó alteración en la intensidad de la señal en la parte mesial de los lóbulos temporales compatible con las descripciones en otras publicaciones35, también se ha reportado compromiso del hipocampo y la amígdala.44 En algunas ocasiones la encefalitis por anticuerpos antiGABAR y AMPAR puede manifestar cambios en los lóbulos temporales en la parte mesial, así como una RMN cerebral normal.45-47 En pacientes con encefalitis límbica paraneoplásica se pueden observar lesiones hiperintensas en T2 uni o bilaterales incluyendo hipocampo.48-50
En la encefalitis límbica las imágenes de tomografía por emisión de positrones (PET) F-fluro-2-deoxy-D-glucosa (FDG) pueden mostrar hipo o hiper metabolismo en los lóbulos temporales, incluso en pacientes con apariencia normal en la RMN.51,52
Más del 75% de pacientes con este síndrome tienen estudio electroencefalográfico anormal, predominantemente con actividad lenta o desorganizada frontotemporal o generalizada (delta-theta) en ausencia de descargas epilépticas.28 En la encefalitis por anticuerpos anti NMDA-R se ha descrito un hallazgo electroencefalográfico específico para este desorden denominado como extreme delta brush53 no observado en nuestros casos, que por lo demás no revelaron hallazgos específicos y predominó el patrón lento difuso sugestivo de encefalopatía.
Respecto al manejo de la encefalitis autoinmune, la FDA no ha aprobado un tratamiento estandarizado, por lo cual nos hemos basado en pautas que incluyen una primera línea con pulsos de esteroides seguidos de altas dosis orales de prednisona, IVIG y plasmaféresis y una segunda con ciclofosfamida y rituximab.6,15 El caso 6 requirió terapias con ciclofosfamida y rituximab no sin antes resolver múltiples inconvenientes administrativos para lograr la autorización de estos medicamentos. Aunque no existe un estudio que demuestre cuál es la terapia ideal en cada caso ni la más eficaz6, en los últimos años hay una tendencia por preferir rituximab, considerado en algunas series como la terapia de elección.54 En nuestra institución hemos tratado de iniciar con pulsos altos de esteroides durante 5 días, en caso de no respuesta el paciente se traslada a la UCI para realizar sesiones de plasmaféresis ya que contamos con amplia experiencia en esta terapia. En promedio se requieren entre 5 y 10 sesiones dependiendo de la severidad del cuadro clínico. En ningún caso hemos usado inmunoglobulina debido a su alto costo y la falta de evidencia que sugiera que es mejor que la plasmaféresis, sin embargo se ha evidenciado buena respuesta con inmunoglobulina en especial en pacientes con anticuerpos antiNMDA.6,27 De las terapias de segunda línea, en general iniciamos con ciclofosfamida debido al menor costo en relación con rituximab y a múltiples limitaciones administrativas que se han generado cuando este último se solicita. Los agentes ahorradores de esteroides como azatioprina y micofenolato tienen pobre evidencia en la eficacia a largo plazo para la prevención de recaídas22, aunque es la terapia inmunomoduladora más usada en el manejo crónico de estos pacientes.
La experiencia en cuanto a los resultados clínicos es variada, en algunas series se ha reportado alta probabilidad de mortalidad y secuelas cognitivas10, mientras otras informan peor pronóstico que las demás formas de encefalitis incluyendo las virales.55 En nuestros casos encontramos una mortalidad del 17% (1 paciente) que presentó severo compromiso hemodinámico y disautonomias con el consecuente fallecimiento. La principal secuela fue la encefalopatía de moderada a severa con alteraciones cognitivas. En general el ingreso a UCI se debe a alteraciones severas del sensorio, necesidad de protección e intubación, estado epiléptico10 y para la realización de plasmaféresis. Aunque nuestra serie es corta, podemos evidenciar que los pacientes requieren hospitalizaciones prolongadas, múltiples estudios de extensión (para descartar diagnósticos diferenciales y definir la asociación con un síndrome paraneoplásico), implican adicionalmente la necesidad de líneas de manejo de alto costo con un pronóstico ominoso debido a la alta refractariedad de la enfermedad. A pesar de las limitaciones con las que cuenta nuestra institución, tenemos ahora en cuenta siempre la importancia de considerar causas autoinmunes en pacientes con un cuadro que sugiera encefalitis, incluso así no sea una presentación típica, tratando en la mayoría de los casos de no retrasar la terapia inmunomoduladora la cual puede iniciarse sin tener los resultados confirmatorios10; así mismo, estamos envía de implementar las diferentes opciones de manejo terapéutico que se recomiendan en forma temprana y escalonada.
CONFLICTO DE INTERESES
Ninguno de los autores recibió alguna remuneración o ayuda financiera para desarrollar el trabajo. No existe ningún conflicto de intereses por parte de alguno de los autores.
Referencias
1. Lancaster E. The Diagnosis and Treatment of Autoimmune Encephalitis. Journal of clinical neurology (Seoul, Korea). 2016;12(1):1-13.
2. Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Annals of neurology. 2014;76(2):168-84.
3. Guan HZ, Ren HT, Cui LY. Autoimmune Encephalitis: An Expanding Frontier of Neuroimmunology. Chinese medical journal. 2016;129(9):1122-7.
4. Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Annals of the New York Academy of Sciences. 2015;1338:94-114.
5. Dubey D, Sawhney A, Greenberg B, Lowden A, Warnack W, Khemani P, et al. The spectrum of autoimmune encephalopathies. Journal of neuroimmunology. 2015;287:93-7.
6. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. The Lancet Neurology. 2016;15(4):391-404.
7. Vollmer TL, McCarthy M. Autoimmune encephalitis: A more treatable tragedy if diagnosed early. Neurology. 2016;86(18):1655-6.
8. Bale JF, Jr. Virus and Immune-Mediated Encephalitides: Epidemiology, Diagnosis, Treatment, and Prevention. Pediatric neurology. 2015;53(1):3-12.
9. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. The Lancet Neurology. 2013;12(2):157-65.
10. Mittal MK, Rabinstein AA, Hocker SE, Pittock SJ, Wijdicks EF, McKeon A. Autoimmune Encephalitis in the ICU: Analysis of Phenotypes, Serologic Findings, and Outcomes. Neurocritical care. 2016;24(2):240-50.
11. Davis R, Dalmau J. Autoimmunity, seizures, and status epilepticus. Epilepsia. 2013;54 Suppl 6:46-9.
12. Sharma A, Dubey D, Sawhney A, Janga K. GAD65 Positive Autoimmune Limbic Encephalitis: A Case Report and Review of Literature. Journal of clinical medicine research. 2012;4(6):424-8.
13. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30(17):5866-75.
14. Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. Journal of neurology. 2010;257(4):509-17.
15. Ramanathan S, Mohammad SS, Brilot F, Dale RC. Autoimmune encephalitis: recent updates and emerging challenges. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2014;21(5):722-30.
16. Vernino S, Geschwind M, Boeve B. Autoimmune encephalopathies. The neurologist. 2007;13(3):140-7.
17. Wingfield T, McHugh C, Vas A, Richardson A, Wilkins E, Bonington A, et al. Autoimmune encephalitis: a case series and comprehensive review of the literature. QJM : monthly journal of the Association of Physicians. 2011;104(11):921-31.
18. Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. Journal of neurology, neurosurgery, and psychiatry. 2012;83(6):638-45.
19. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. The Lancet Neurology. 2008;7(12):1091-8.
20. Day GS, High SM, Cot B, Tang-Wai DF. Anti-NMDA-receptor encephalitis: case report and literature review of an under-recognized condition. Journal of general internal medicine. 2011;26(7):811-6.
21. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. The Lancet Neurology. 2011;10(1):63-74.
22. Irani SR, Vincent A. NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep. 2011;11(3):298-304.
23. Zandi MS, Irani SR, Follows G, Moody AM, Molyneux P, Vincent A. Limbic encephalitis associated with antibodies to the NMDA receptor in Hodgkin lymphoma. Neurology. 2009;73(23):2039-40.
24. Iizuka T, Hara A. [Anti-NMDA receptor antibody-mediated encephalitis/encephalopathy]. Rinsho byori The Japanese journal of clinical pathology. 2009;57(3):252-61.
25. Kleinig TJ, Thompson PD, Matar W, Duggins A, Kimber TE, Morris JG, et al. The distinctive movement disorder of ovarian teratoma-associated encephalitis. Movement disorders : official journal of the Movement Disorder Society. 2008;23(9):1256-61.
26. Smith JH, Dhamija R, Moseley BD, Sandroni P, Lucchinetti CF, Lennon VA, et al. N-methyl-D-aspartate receptor autoimmune encephalitis presenting with opsoclonus-myoclonus: treatment response to plasmapheresis. Archives of neurology. 2011;68(8):1069-72.
27. Sansing LH, Tuzun E, Ko MW, Baccon J, Lynch DR, Dalmau J. A patient with encephalitis associated with NMDA receptor antibodies. Nature clinical practice Neurology. 2007;3(5):291-6.
28. Florance NR, Davis RL, Lam C, Szperka C, Zhou L, Ahmad S, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Annals of neurology. 2009;66(1):11-8.
29. Gabilondo I, Saiz A, Galan L, Gonzalez V, Jadraque R, Sabater L, et al. Analysis of relapses in anti-NMDAR encephalitis. Neurology. 2011;77(10):996-9.
30. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. The Lancet Neurology. 2010;9(8):776-85.
31. Barber PA, Anderson NE, Vincent A. Morvan's syndrome associated with voltage-gated K+ channel antibodies. Neurology. 2000;54(3):771-2.
32. Irani S, Lang B. Autoantibody-mediated disorders of the central nervous system. Autoimmunity. 2008;41(1):55-65.
33. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain : a journal of neurology. 2010;133(9):2734-48.
34. Striano P. Faciobrachial dystonic attacks: seizures or movement disorder? Annals of neurology. 2011;70(1):179-80; author reply 80.
35. Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain : a journal of neurology. 2004;127(Pt 3):701-12.
36. Buckley C, Oger J, Clover L, Tuzun E, Carpenter K, Jackson M, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Annals of neurology. 2001;50(1):73-8.
37. Pozo-Rosich P, Clover L, Saiz A, Vincent A, Graus F. Voltage-gated potassium channel antibodies in limbic encephalitis. Annals of neurology. 2003;54(4):530-3.
38. Jarius S, Hoffmann LA, Stich O, Clover L, Rauer S, Vincent A, et al. Relative frequency of VGKC and 'classical' paraneoplastic antibodies in patients with limbic encephalitis. Journal of neurology. 2008;255(7):1100-1.
39. Prosser HM, Gill CH, Hirst WD, Grau E, Robbins M, Calver A, et al. Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Molecular and cellular neurosciences. 2001;17(6):1059-70.
40. Heine J, Pruss H, Bartsch T, Ploner CJ, Paul F, Finke C. Imaging of autoimmune encephalitis--Relevance for clinical practice and hippocampal function. Neuroscience. 2015;309:68-83.
41. Wegner F, Wilke F, Raab P, Tayeb SB, Boeck AL, Haense C, et al. Anti-leucine rich glioma inactivated 1 protein and anti-N-methyl-D-aspartate receptor encephalitis show distinct patterns of brain glucose metabolism in 18F-fluoro-2-deoxy-d-glucose positron emission tomography. BMC neurology. 2014;14:136.
42. Tojo K, Nitta K, Ishii W, Sekijima Y, Morita H, Takahashi Y, et al. A Young Man with Anti-NMDAR Encephalitis following Guillain-Barre Syndrome. Case reports in neurology. 2011;3(1):7-13.
43. Maqbool M, Oleske DA, Huq AH, Salman BA, Khodabakhsh K, Chugani HT. Novel FDG-PET findings in anti-NMDA receptor encephalitis: a case based report. Journal of child neurology. 2011;26(10):1325-8.
44. Kotsenas AL, Watson RE, Pittock SJ, Britton JW, Hoye SL, Quek AM, et al. MRI findings in autoimmune voltage-gated potassium channel complex encephalitis with seizures: one potential etiology for mesial temporal sclerosis. AJNR American journal of neuroradiology. 2014;35(1):84-9.
45. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Annals of neurology. 2009;65(4):424-34.
46. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. The Lancet Neurology. 2010;9(1):67-76.
47. Boronat A, Sabater L, Saiz A, Dalmau J, Graus F. GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology. 2011;76(9):795-800.
48. Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain : a journal of neurology. 2000;123 ( Pt 7):1481-94.
49. Lawn ND, Westmoreland BF, Kiely MJ, Lennon VA, Vernino S. Clinical, magnetic resonance imaging, and electroencephalographic findings in paraneoplastic limbic encephalitis. Mayo Clinic proceedings. 2003;78(11):1363-8.
50. Ances BM, Vitaliani R, Taylor RA, Liebeskind DS, Voloschin A, Houghton DJ, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain : a journal of neurology. 2005;128(Pt 8):1764-77.
51. Scheid R, Lincke T, Voltz R, von Cramon DY, Sabri O. Serial 18F-fluoro-2-deoxy-D-glucose positron emission tomography and magnetic resonance imaging of paraneoplastic limbic encephalitis. Archives of neurology. 2004;61(11):1785-9.
52. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. 2008;10(3):131-7.
53. Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. 2012;79(11):1094-100.
54. Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, Murphy TK, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain : a journal of neurology. 2012;135(Pt 11):3453-68.
55. Thakur KT, Motta M, Asemota AO, Kirsch HL, Benavides DR, Schneider EB, et al. Predictors of outcome in acute encephalitis. Neurology. 2013;81(9):793-800.