REPORTE DE CASO
DISPLASIA ARRITMOGÉNICA DEL VENTRÍCULO DERECHO
ARRHYTHMOGENIC RIGHT VENTRICULAR DYSPLASIA
Fecha recibido: octubre 23 de 2017
Fecha aceptado: junio 10 de 2018
Juan José Chavesa, Caterin Niñob, Adriana Bryon MDc, Hernán Medina MDd, Francisco Calle MDd, José Fernando Polo MDe, Rafael Parra-Medina MDf
Correo electrónico autor principal Sr. Juan José Chaves: juan-chavescabezas@hotmail.com
a Facultad de Medicina, Fundación Universitaria de Ciencias de la Salud, Bogotá DC, Colombia.
b Medicina Interna, Fundación Universitaria de Ciencias de la Salud, Bogotá DC, Colombia.
c Patología, Universidad Militar Central, Bogotá DC, Colombia.
d Patología, Instituto Nacional de Medicina Legal. Bogotá DC, Colombia.
e Servicio de Patología, Hospital de San José, Hospital Infantil Universitario de San José, Fundación Universitaria de Ciencias de la Salud. Bogotá DC, Colombia.
f Servicio de Patología, Hospital de San José, Fundación Universitaria de Ciencias de la Salud. Bogotá DC, Colombia.
Resumen
La displasia arritmogénica del ventrículo derecho (DAVD) es una cardiomiopatía caracterizada por el reemplazo de miocitos por tejido fibroadiposo con herencia autosómica dominante. Ocupa el segundo lugar como muerte súbita en adultos jóvenes y es causante de un gran porcentaje en atletas. Su clínica es variable, debido a que puede presentarse en reposo o luego de actividad física con síntomas inespecíficos como palpitaciones, síncope y dolor torácico. Se presentan tres casos autópsicos de diferentes instituciones cuya manifestación clínica fue muerte súbita.
Palabras clave: displasia arritmogénica, ventrículo, arritmia, muerte súbita.
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS. Este es un artículo Open Access bajo la licencia CC BY-NC-ND (http:// creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
Arrhythmogenic right ventricular dysplasia (ARVD) is a cardiomyopathy inherited in an autosomal dominant pattern characterized by replacement of myocytes with fibrofatty tissue. ARVD is the second cause of sudden death in young adults and accounts for a great proportion of deaths in athletes. Clinical presentation is variable, it may occur during rest or after physical activity involving unspecific symptoms such as palpitations, syncope and chest pain. We here report three autopsy cases, referred from various healthcare institutions, in which sudden death was the first manifestation.
Key Words: Arrhythmogenic dysplasia, ventricle, arrhythmia, sudden death
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS.This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Introducción
La displasia arritmogénica del ventrículo derecho (DAVD) es una cardiomiopatía caracterizada por el reemplazo de miocitos por tejido fibroadiposo con herencia autosómica dominante.1 Cursa con defectos en proteínas desmosomales desencadenando arritmias ventriculares no isquémicas originadas en el ventrículo derecho.2,3 Las proteínas que en mayor medida se ven afectadas son la placoglobina, desmoplaquina, placofilina-2, desmocolina-2 y desmogleina-2.4
Es la segunda causa de muerte súbita en adultos jóvenes (2-5%) y de un gran porcentaje en atletas.2 Su clínica es variable debido a que puede presentarse en reposo o luego de actividad física con síntomas inespecíficos como palpitaciones, sincope y dolor torácico.1
La prevalencia varía entre 1:2500 a 1:50001,2, siendo un dato impreciso debido al gran número de casos no diagnosticados2, lo cual se debe a que su diagnóstico es difícil por la presentación clínica y a veces con datos insatisfactorios en la biopsia debido a que el ventrículo derecho es delgado y su componente subepicárdico es difícil de apreciar en estudios de imagen siendo con frecuencia solo investigados en la necropsia clínica.1
Presentamos tres casos de DAVD que debutan con muerte súbita cardiaca, dos pacientes tenían historia clínica de arritmia cardiaca. Fueron analizados en tres instituciones diferentes de Bogotá, Colombia (Hospital de San José, Hospital Infantil Universitario de San José y el Instituto de Medicina Legal y Ciencias Forenses).
Presentación de casos
Caso Nº 1
Mujer de 32 años con antecedentes de hipotiroidismo y arritmia cardiaca. Ingresa al servicio de urgencias por pérdida súbita del estado de conciencia de 30 minutos de evolución. Al examen físico se determina un Glasgow de 3/15, sin signos vitales, pupilas midriáticas no reactivas, sin respuesta a estímulos dolorosos, con glucometria de 130 mg/dl y visoscopio que evidencia fibrilación ventricular sin pulso. Se realiza reanimación cardiovascular (RCP) avanzada por 35 minutos sin obtener respuesta.
Según datos de la historia clínica la paciente tenia diagnóstico por holter de arritmia ventricular Lown y Wolf tipo IVB y arritmia supraventricular de bajo grado; en el ecocardiograma se evidenció derrame pericárdico leve sin compromiso hemodinámico con numerosas extrasístoles supraventriculares durante el procedimiento con buena función sistólica biventricular (FE 55%).
Caso Nº 2
Paciente masculino de 28 años de edad que ingresa al servicio de urgencias por presentar cuadro de sensación vertiginosa asociada con dolor torácico opresivo, el electrocardiograma reporta taquicardia supraventricular no especificada paroxística con bloqueo de rama derecha. El paciente presenta dolor torácico intenso con cianosis central y posterior perdido de la conciencia. Se realizó RCP avanzada sin obtener respuesta.
Caso N º 3
Mujer de 43 años sin antecedentes personales que ingresa a urgencias con cuadro clínico generalizado de 12 horas de evolución de dolor abdominal tipo cólico que se irradiaba a región dorsal, vómito, dificultad respiratoria súbita y pérdida del conocimiento. Se realizó RCP avanzada sin obtener respuesta.
Hallazgos autópsicos
En los tres casos el único hallazgo relevante fue la cardiomegalia, con pesos de 355 g, 380 g y 385 g. No se reconocieron otras alteraciones morfológicas (figura 1).
En el estudio histopatológico de los tres casos se observó una banda fibrosa subepicárdica gruesa, cambio a tejido adiposo que remplaza la pared del miocardio en todo el espesor con una cantidad de miocitos residuales menor del 60% (figura 2).
Discusión
La DAVD es una cardiomiopatía hereditaria caracterizada por atrofia miocárdica, reemplazo fibroadiposo y adelgazamiento de la pared con dilatación y formación de aneurismas, cuya clínica es muy variable. Puede ir desde una fase asintomática hasta arritmias ventriculares, disfunción ventricular derecha y muerte súbita. Aunque es una patología del ventrículo derecho, es también vista en el izquierdo. 5
La primera descripción fue en 1736 por Giovanni Lancisi, quien en su libro “Motu Cordis et Aneurysmatibus” reportó una familia con enfermedad recurrente en cuatro generaciones donde los afectados presentaban palpitaciones, falla cardiaca, dilatación y aneurismas en el ventrículo derecho.2 Luego Marcus en 1982 reportó la enfermedad en adultos siendo el primero que enfatizó el origen de las arritmias en el ventrículo derecho mostrando que su sustrato histopatológico era el reemplazo fibroadiposo en la pared libre del ventrículo derecho.6
Aunque aún la patogenia exacta de la enfermedad es desconocida, las teorías más aceptadas son las que afirman que la etiología es genética, asociada a menudo con un patrón de herencia autosómica dominante de penetrancia incompleta y de expresividad variable.1 Algunos estudios describen otro de herencia autosómica recesiva como parte de síndromes cardiocutáneos como el síndrome de Carvajal.7
Su base genética está dada por la mutación en genes de los desmosomas, donde una persona con una mutación tiene la probabilidad de 30 a 50% de desarrollar la patología, pero la presencia de más de una mutación aumenta el riesgo.1 Se ha identificado la mutación en proteínas desmosomales hasta en 60% de los pacientes con DAVD4, siendo la placoglobina, desmoplaquina, placofilina-2, desmocolina-2 y desmogleina-2 las proteínas que más se afectan.4 Por lo tanto, la falta de la mutación en una proteína no excluye la enfermedad.8
En general, la función de los desmosomas es formar puentes entre células y filamentos intermedios o entre células cardiacas, siendo importantes también en la transducción de señales vía Wnt-BetaCatenina.1 A partir de su función se tienen dos modelos de generación de la displasia arritmogénica del ventrículo derecho: el de degeneracion-inflamacion (MDI) y el modelo de transdiferenciacion (MT).1 En MDI se observa que el daño es el resultado de las fuerzas mecánicas que alteran la arquitectura cardiaca, donde desmosomas disfuncionales son incapaces de soportar la presión, dando como resultado el deterioro de las células generando necrosis, inflamación y finalmente reemplazo fibroso.1 En MT, se afirma que cuando los desmosomas son disfuncionales hay un aumento en la placoglobina que se traslada al núcleo inhibiendo la señalización Wnt, causando un cambio de expresión celular de cardiogénico a adipogénico.1 A pesar de esto, algunos estudios en familias han mostrado que las mutaciones desmosomales están presentes en menos del 30%.9 Otras teorías afirman que la etiología es inflamatoria, debido a que se ha evidenciado presencia de virus cardiotrópicos en los casos con inflamación miocárdica pudiendo ser importantes en el origen de las taquiarritmias.2,3 De igual manera puede tener también origen inmunitario por la presencia de anticuerpos antimiocárdicos (M2 y MHC) en algunos casos.3
El diagnóstico clínico es complejo debido a que el paciente puede presentar una fase subclínica con anormalidades estructurales ocultas donde no hay síntomas y el ataque cardiaco puede ser la primera y única manifestación; o estar en una fase de desórdenes eléctricos acompañada de palpitaciones y síncope que terminará en una fase de falla ventricular derecha o biventricular.1,2. Los hallazgos electrocardiográficos son muy variables y poco específicos, siendo las arritmias ventriculares las más comunes, van desde taquicardia ventricular sostenida hasta fibrilación ventricular con posterior paro cardiaco.10 La morfología del complejo QRS y el eje de la taquicardia ventricular reflejan su origen. Se pueden evidenciar ondas T invertidas en derivaciones derechas, amplificación del complejo QRS y ondas épsilon que sugieren áreas de conducción interventricular lenta debido a las islas de grasa que representan un obstáculo para la conducción eléctrica del miocardio.2
En la actualidad los criterios de la Task Force modificados (2010) son los más utilizados para hacer el diagnóstico de DAVD, siendo un sistema de puntuación con criterios mayores y menores basados en la evidencia de disfunciones ventriculares electrocardiográficas o estructurales, en la historia familiar y en las pruebas genéticas. Otras herramientas son la resonancia magnética con gadolinio y mapeo tridimensional electroanatómico, que han demostrado aumentar la sensibilidad del diagnóstico.2
En el estudio macroscópico se debe tener en cuenta que los cambios estructurales pueden ser limitados o ausentes, lo cual hace difícil el diagnóstico en la necropsia.4 Además, la infiltración adiposa no es patognomónica de la enfermedad, ya que es común su presencia en regiones anterolaterales y apicales del ventrículo derecho en personas sin cardiopatía2, y con mayor porcentaje (50%) en personas con edad avanzada, alcohólicas y obesas.1 El fenómeno de reemplazo es progresivo en el tiempo desde epicardio a endocardio, generando en la pared libre del ventrículo dilatación y aneurismas.2
En los hallazgos histopatológicos se debe tener en cuenta que para el diagnóstico se necesita que la cantidad de miocitos residuales sea menor del 60% y cambio fibroso en la pared libre del ventrículo derecho, con o sin reemplazo adiposo.4 Esta disminución de la cantidad de miocitos se da por una destrucción celular a cargo de un infiltrado compuesto de linfocitos y macrófagos.1 Los estudios muestran que para que la biopsia sea útil debe tomarse del “triángulo de displasia”, el cual es el ápex, infundíbulo y pared posteroinferior, recalcando que las muestras de la pared libre del ventrículo derecho en vez de las del septum puede generar mayor sensibilidad en el diagnóstico.1 Como ayudas confirmatorias, la tinción de tricrómico de Masson puede ayudar a diferenciar áreas de fibrosis5 y la microscopía electrónica puede evidenciar el remodelado de los discos intercalares.1
En los últimos años se ha sugerido el estudio inmunohistoquímico para placoglobina como nuevo a prueba diagnóstica, mediante lo cual se ha evidenciado en diferentes estudios que hay una disminución de la expresión de esta proteína entre 68 y 85%4,11, recalcando que ningún paciente con mutaciones desmosomales tiene niveles de placoglobina normales.11 De igual manera este hallazgo no es patognomónico de la displasia, presentándose también en patologías como cardiomiopatía dilatada y de Takotsubo.11
Se debe tener en cuenta que más de la mitad de estudios posmortem, donde algunos autores reportan hasta 93%9, hay compromiso del ventrículo izquierdo limitado al subepicardio de la pared posterolateral2, evidenciándose por técnicas de imagen que el compromiso izquierdo es típico desde los inicios de la enfermedad.9
El desfibrilador automático implantable y el empleo de fármacos antiarrítmicos han demostrado eficacia en pacientes con factores de mal pronóstico, que incluyen corta edad de presentación, antecedentes familiares de muerte súbita juvenil, amplitud del QRS ≥ 40 ms, inversión de la onda T, participación del ventrículo izquierdo, antecedente de sincope y taquicardia ventricular o paro cardiaco.10 En pacientes con bajo riesgo o asintomáticos se recomienda terapia farmacológica con betabloqueadores.8 Cuando hay falla terapéutica en caso de arritmias o insuficiencia cardiaca congestiva, el trasplante cardiaco es considerado la única opción terapéutica.2
Por último, podemos decir que la DAVD es una entidad de difícil diagnóstico, que al parecer es más frecuente de lo que se aprecia, cada vez con mayor número de casos reportados.1 A menudo se identifica en la necropsia debido a su gran variedad clínica y algunas veces los datos insatisfactorios de estudios diagnósticos complementarios llevan a un diagnóstico inapropiado o tardío.
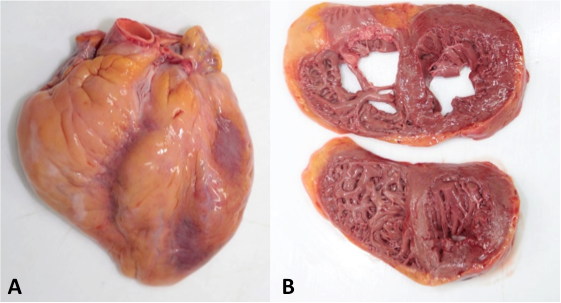
Figura 1. Fotos del caso Nº 3. A. Cardiomegalia con peso de 385 g. B. Espesor de la pared ventricular izquierda de 1.4 cm y la derecha 0.2 cm.
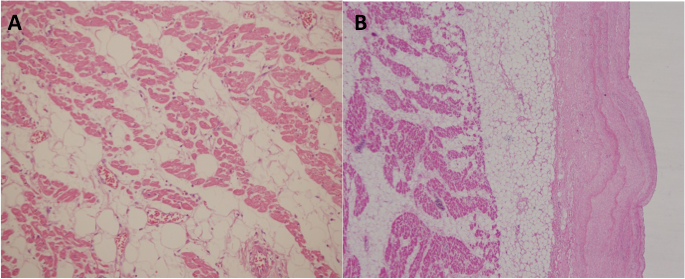
Figura 2. A. Tejido adiposo que remplaza la pared del miocardio en todo el espesor con una cantidad de miocitos residuales menor del 60%. H&E 10x. B. Presencia de fibrosis subepicárdica H&E 10x.
DECLARACIÓN DE CONFLICTO DE INTERÉS
Los autores declaran no tener conflictos de interés en relación a este estudio.
Referencias
1. McGregor SM, Husain AN. A Brief Review and Update of the Clinicopathologic Diagnosis of Arrhythmogenic Cardiomyopathy. Archives of pathology & laboratory medicine. 2015;139(9):1181-6. Epub 2015/09/01.
2. Thiene G, Corrado D, Basso C. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet journal of rare diseases. 2007;2:45. Epub 2007/11/16.
3. Wei J, Tang J, Xia L, Chen X, Wang DW. A case of arrhythmogenic right ventricular cardiomyopathy without arrhythmias. Diagnostic pathology. 2012;7:67. Epub 2012/06/14.
4. Munkholm J, Andersen CB, Ottesen GL. Plakoglobin: a diagnostic marker of arrhythmogenic right ventricular cardiomyopathy in forensic pathology? Forensic science, medicine, and pathology. 2015;11(1):47-52. Epub 2015/01/01.
5. Kondo T. A case of arrhythmogenic right ventricular cardiomyopathy (ARVC/D) in which tenascin C immunostaining made the assessment of myocardial remodeling possible. Soudni lekarstvi. 2014;59(3):24-5. Epub 2014/09/05.
6. Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384-98. Epub 1982/02/01.
7. Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology. 2004;13(4):185-94. Epub 2004/06/24.
8. Orgeron GM, Calkins H. Advances in the Diagnosis and Management of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Current cardiology reports. 2016;18(6):53. Epub 2016/04/25.
9. Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115(13):1710-20. Epub 2007/03/21.
10. Ermakov S, Scheinman M. Arrhythmogenic Right Ventricular Cardiomyopathy - Antiarrhythmic Therapy. Arrhythmia & electrophysiology review. 2015;4(2):86-9. Epub 2016/02/03.
11. Munkholm J, Christensen AH, Svendsen JH, Andersen CB. Usefulness of immunostaining for plakoglobin as a diagnostic marker of arrhythmogenic right ventricular cardiomyopathy. The American journal of cardiology. 2012;109(2):272-5. Epub 2011/11/01.