REPORTE DE CASO / CASE REPORT
ENCEFALITIS DE RASMUSSEN: DIAGNÓSTICO Y ENFOQUE TERAPÉUTICO
RASMUSSEN ENCEPHALITIS: DIAGNOSTIC AND THERAPEUTIC APPROACHES
Fecha recibido: febrero 26 de 2018
Fecha aceptado: septiembre 12 de 2018
Eugenia Espinosa MDa, Angélica Fuentes MDa, Amanda Liliana Naranjo MDb
Correo electrónico autora principal: Dra. Eugenia Espinosa: eugeniaespinosa@yahoo.com
a Neurología Pediátrica. Universidad Militar Nueva Granada Bogotá D.C, Colombia
b Neurología, Fundación Universitaria Ciencias de la Salud, Bogotá DC, Colombia.
Resumen
Objetivo: describir el caso de una paciente escolar con diagnóstico de encefalitis de Rasmussen de presentación aguda y el enfoque terapéutico. Presentación: paciente femenina de 5 años y nueve meses que cursa con cuadro de evolución progresiva caracterizado por posturas distónicas que se inicia en pierna izquierda seguida de miembro superior ipsilateral asociadas con crisis focales con semiología de inicio opercular que la conducen a estatus epiléptico. La resonancia magnética cerebral evidencia cambios estructurales progresivos con atrofia hemisférica y del núcleo basal derechos, se inicia terapia antiepiléptica e inmunomoduladora con buena respuesta. Discusión: se realizó una revisión de la literatura sobre la presentación clínica, diagnóstico y mejores opciones terapéuticas. Conclusiones: se trata de una escolar con síndrome de Rasmussen en etapa aguda, con adecuada evolución clínica luego de inicio de inmunoterapia. En Colombia existen pocos reportes, nuestro propósito es realizar la presentación de un caso y revisar el enfoque diagnóstico y terapéutico.
Palabras clave: encefalitis Rasmussen, síndrome Rasmussen, epilepsia parcial continua, inmunoduladores, tratamiento.
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS. Este es un artículo Open Access bajo la licencia CC BY-NC-ND (http:// creativecommons.org/licenses/by-nc-nd/4.0/).
Abstract
Objective: to describe the case of a school-age patient aged 5 years 9 months diagnosed with acute Rasmussen encephalitis (RE), and its therapeutic approach. Case presentation: a girl aged 5 years 9 months presenting with progressive dystonic postures starting in the left lower limb with spread to the ipsilateral upper limb associated with focal seizures characterized by opercular epilepsy semiology which lead to status epilepticus. Cerebral magnetic resonance imaging revealed progressive structural changes with atrophy of the right cerebral hemisphere and basal nuclei. Anti-epileptic and immunmodulator therapy was initiated obtaining good response. Discussion: a review of the literature on RE clinical presentation, diagnosis and best treatment options was conducted. Conclusions: this is the case of a young girl with Rasmussen syndrome in the acute stage evidencing proper clinical progression after receiving immunotherapy. Few reports exist on this topic in Colombia. Our purpose was to present a case and review RE diagnostic and therapeutic approaches.
Key Words: Rasmussen encephalitis, Rasmussen syndrome, epilepsia partialis continua, immunomodulator, treatment
© 2018 Fundación Universitaria de Ciencias de la Salud - FUCS. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Introducción
En 1958 Theodore Rasmussen y col. del Instituto Neurológico de Montreal informaron de tres pacientes con crisis focales debido a una encefalitis crónica localizada. Desde finales de 1980 la mayoría de los investigadores y clínicos adoptaron el término de encefalitis de Rasmussen o síndrome de Rasmussen para esta condición.1 A partir de esta fecha se ha identificado un amplio espectro de características clínicas que sumado a la falta de comprensión exacta acerca de su etiología, ha generado dilemas en la toma de decisiones clínicas.2 Es un trastorno poco frecuente, que se presenta con una incidencia de 2-4 casos por 10 millones de personas menores de 18 años y una prevalencia de 0-8 por 100.000 personas; sin predominio de género, geográfico ni étnico. Se presenta a cualquier edad desde la infancia hasta la adolescencia con un pico a los 6 años y el 10% de casos ocurren en adolescentes y adultos.2 Consiste en un trastorno neurológico crónico, caracterizado por epilepsia farmacorresistente, déficit neurológico y deterioro cognitivo progresivo debido a inflamación unilateral de la corteza cerebral. Este curso de la inflamación es consistente con una enfermedad de mediación inmune, sin identificar siempre marcadores serológicos o intratecales.3 En Colombia existen pocos casos reportados, nuestro propósito es realizar la presentación de un caso y revisar el enfoque diagnóstico y terapéutico.
Presentación del caso
Paciente escolar de sexo femenino de 5 años y 9 meses con cuadro clínico de carácter progresivo que se inicia con movimientos involuntarios no estereotipados sin sentido en miembro inferior izquierdo en dirección anteroposterior, de forma intermitente sin factor precipitante, que desaparecen durante el sueño, seguidos por perdida de la fuerza en la misma extremidad lo que condiciona inestabilidad y caídas frecuentes, que luego compromete miembro superior ipsilateral; se asocian cambios en el comportamiento dados por irritabilidad, episodios de agresividad y labilidad emocional. En los últimos 2 meses se presentaron episodios de sialorrea, detención de la actividad motora, fijación de la mirada, seguidos de sacudidas palpebrales en ojo izquierdo de
segundos de duración, con una frecuencia de cinco veces al día, ocurriendo tanto en vigilia como en sueño, sin compromiso del estado de conciencia.
Paciente previamente sana, producto de segunda gestación, embarazo controlado a término, parto por vía vaginal, con adecuadas medidas antropométricas y adaptación espontanea; sin antecedentes patológicos, neurodesarrollo con hitos logrados a las edades correspondientes, en la actualidad escolarizada en jardín infantil, con buen desempeño escolar y sin antecedentes familiares de enfermedad neurológica o sistémica. Al examen neurológico no hay compromiso de funciones mentales ni déficit cognitivo y como hallazgos positivos se encuentran hemianopsia temporal izquierda, postura distónica en flexión de miembro superior izquierdo y hemiparesia ipsilateral con fuerza estimada en 4/5, reflejos miotendinosos asimétricos exaltados en hemicuerpo izquierdo, sin signos cerebelosos.
En consultas previas al ingreso le realizaron estudios con perfil autoinmune: C3-C4, C ancas y P ancas, anti-Ro, anti-La, Anti-SM y anti-RNP. Los anticuerpos antifosfolípidos y el perfil metabólico sin hallazgos positivos, en el líquido cefalorraquídeo en el análisis citoquímico, gram y cultivo no revelaron alteraciones, PCR para herpes virus negativos. ANAS positivos con patrón moteado 1:160 y estudio de bandas oligoclonales positivo en LCR y negativo a nivel sérico.
Neuroimágenes
En la RMN simple de cerebro (figuras 1y 2) se aprecian cambios malácicos cortico-subcorticales que afectan el lóbulo frontal derecho en especial los giros frontal medio e inferior, con adelgazamiento de la corteza y efecto retráctil sobre el cuerno anterior del ventrículo lateral del mismo lado y discreta hiperintensidad en la sustancia blanca periventricular y en centros semiovales visualizados en T2 y FLAIR. Una segunda imagen al mes siguiente la resonancia simple y contrastada de cerebro muestra disminución difusa del volumen central cortical del lóbulo frontal derecho sin alteraciones focales y la última a los 4 meses se observa atrofia frontoparietoinsular además de gliosis frontal y de núcleo basal derecho. Videoelectroencefalograma interictal (figura 3) con actividad epileptiforme de tipo ondas agudas en la región fronta l izquierda, durante las maniobras de activación en la hiperventilación se observa lentificación difusa de la actividad cerebral de fondo. Durante el estudio se registran múltiples crisis que ocurren durante el sueño y vigilia, la paciente presenta sialorrea, fijación de mirada, clonias de hemicuerpo izquierdo y parpadeo derecho, no hay desconexión con el medio, el trazado eléctrico muestra ondas lentas sobre la región frontal izquierda que difunden a regiones homólogas contralaterales pero no se generalizan.
Con los hallazgos clínicos, eléctricos e imagenológicos descritos, se diagnostica encefalitis de Rasmussen, iniciándose terapia inmunomoduladora y antiepiléptica con oxcarbazepina. Durante su evolución la paciente presenta estado epiléptico refractario requiriendo estancia hospitalaria prolongada y a su egreso se adiciona levetiracetam logrando adecuado control de las crisis, sin mayor deterioro funcional ni cognitivo.
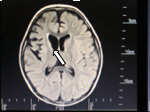
Figura 1. Leve dilatación del cuerno frontal del
ventrículo lateral derecho.
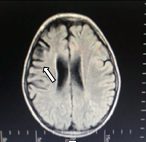
Figura 2. Disminución del volumen parenquimatoso que afecta el lóbulo frontal (flecha).
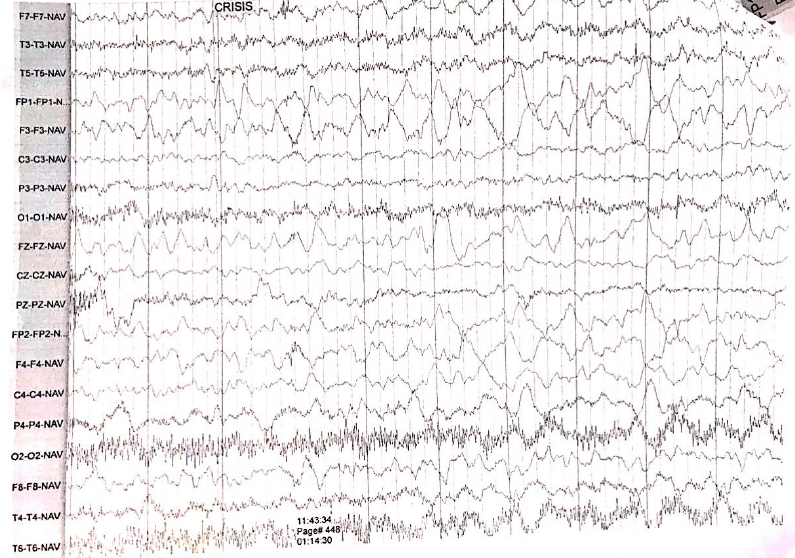
Figura 3. Grafoelementos de morfología epileptiforme de tipo ondas en región frontal derecha.
Discusión
La encefalitis de Rasmussen se describe como una encefalopatía infantil secundaria a inflamación unilateral de la corteza cerebral con una supuesta base fisiopatológica mediada por mecanismos inmunes. Estos han sido reconocidos como responsables de la degeneración del sistema nervioso central y se caracterizan en tres tipos: citotoxicidad por células T, mediada por anticuerpos e inducida por microglia.2 Con relación a los anticuerpos, se han identificado a lo largo de la historia varios autoanticuerpos contra diferentes epítopes antigénicos, cuyo papel en la patogénesis aún no está claro. Dentro de estos se han identificado contra la subunidad 3 del receptor inotrópico de glutamato (iGluR3), los cuales llevan a muerte celular neuronal secundaria a excitotoxicidad.1,4
En la presentación clínica se reconocen tres etapas, un periodo prodrómico en el que ocurre hemiparesia leve y/o crisis infrecuentes, la cual puede preceder a la etapa aguda con un promedio de 7.1 meses, pero puede durar años. Después sigue la etapa aguda, que se presenta en una tercera parte de los pacientes, se caracteriza por convulsiones focales frecuentes. Alrededor del 50% desarrollan epilepsia parcial continua y a medida que la enfermedad avanza aparecen crisis focales de diferente semiología siendo las más frecuentes las focales motoras, encontradas en 77% de los casos, que pueden extenderse a ser bilateral en el 42%, 19% cursan con automatismos, 24% se originan en el área motora suplementaria y 21% en la corteza somatosensorial; además en el trascurso del cuadro clínico las crisis cursan con refractariedad.1-3,5,6
En nuestro caso las características semiológicas ictales corresponden a crisis focales simples continuas con refractariedad a la terapia inicial; se ha descrito en la evolución clínica que en promedio al año siguiente al inicio de la epilepsia estos pacientes presentan hemiparesia, hemianopsia y trastorno del movimiento unilateral. En la actualidad la paciente descrita tiene un importante compromiso motor unilateral izquierdo con alteración en la marcha y postura distónica; la disfasia es otro componente del cuadro clínico si el hemisferio comprometido es el dominante. Las alteraciones comportamentales por lo general preceden a dificultades en la memoria, la atención y el aprendizaje. En la mayoría de los pacientes la progresión del compromiso cognitivo parece correlacionarse con la gravedad de la epilepsia.1-3,5-7
En nuestro caso al inicio de la enfermedad sus padres refieren cambios comportamentales dados por irritabilidad, labilidad emocional e intolerancia, que han mejorado de forma parcial sin observar hasta el momento dificultades escolares ni regresión del desarrollo o de habilidades escolares.
Pocos meses luego del inicio de la etapa aguda, la mayoría de los pacientes muestran un agrandamiento unilateral del sistema ventricular. En T2 y FLAIR se observa hiperintensidad a nivel cortical o subcortical, siendo la región perisilviana el sitio de predilección para cambio de la señal y pérdida de volumen2, como se observa en los estudios de resonancia tomados en la fase temprana en el caso de la paciente descrita. La atrofia cortical ipsilateral de la cabeza del núcleo caudado acompañando la atrofia hemisférica suele ser un signo temprano.2,8 Los estudios imagenológicos del caso nos permitieron observar cambios en la señal de la sustancia blanca periventricular y centros semiovales en la fase inicial, con pérdida de volumen y luego atrofia de localización en el lóbulo frontal, con posterior extensión frontoparietoinsular y a ganglio basal derecho, que se correlaciona con el cuadro clínico de la paciente y los diferentes reportes de casos en otros centros médicos.
En estudios de medición volumétrica se describe una mayor tasa de pérdida de volumen en los primeros 8 meses de la enfermedad durante la fase clínica aguda, con un predominio del compromiso del putamen en lugar del caudado, a nivel de los ganglios basales. Alguna atrofia es siempre evidente, incluso del hemisferio no afectado, probablemente como resultado de la degeneración de las fibras comisurales.2,8
En estudios complementarios como el electroencefalograma (EEG) no se describen hallazgos específicos, sin embargo a partir de un EEG normal al inicio suele aparecer actividad delta de alta amplitud persistente sobre el hemisferio afectado dentro de los meses siguientes del comienzo de las convulsiones.2
Las anormalidades epileptiformes son frecuentes y a menudo se convierten en crisis electrográficas, pero la epilepsia parcial continua no siempre va acompañada de una actividad de superficie ictal reconocible.2 Las anormalidades interictales independientes sobre el hemisferio no afectado emergen dentro de los 6 meses en 25% de los pacientes y en 62% entre 3 y 5 años desde el inicio de las crisis; estas alteraciones contralaterales están asociadas y pueden ser un marcador del deterioro cognitivo pero no de compromiso bilateral.2 Al inicio de la enfermedad nuestra paciente no mostró cambios eléctricos, sin embargo al momento del estatus epiléptico en la monitorización electroencefalográfica fue evidente el compromiso contralateral que se relaciona con lo que se ha observado en otros casos de la misma edad y con igual diagnóstico.
El estudio de líquido cefalorraquídeo (LCR) a pesar de casi siempre aporta poca información, debe llevarse a cabo para excluir infecciones del sistema nervioso central u otros trastornos. Las bandas oligoclonales en el LCR se describen en cerca de la mitad de los pacientes, fue positivo en el presente caso aunque de carácter inespecífico7, lo que puede ser un factor confusional que obliga a descartar otras etiologías autoinmunes.
El consenso Europeo propuso unos criterios diagnósticos en 2005 los cuales siguen siendo aceptados y utilizados en la actualidad. Se requieren tres cardinales para realizar el diagnóstico, los cuales se describen a continuación.1-3
Criterios diagnósticos
En nuestro caso se cumplen los tres criterios principales. Sin embargo ante la falta de algunos de ellos, el diagnóstico se puede establecer con dos de los siguientes criterios alternativos:
1. Clínico: epilepsia parcial continua o déficits cortical unilateral progresivo.
2. Resonancia magnética: atrofia cortical focal hemisférica progresiva.
3. Histopatología: encefalitis mediada por células T con células de la microglia activadas, pero no siempre formando nódulos y astrogliosis reactiva.
El tratamiento va dirigido a reducir la severidad y frecuencia de las crisis; los estudios reportan el manejo durante la fase aguda basado en inmunoterapia y medicamentos antiepilépticos, los cuales a menudo muestran al principio respuesta temporal e impredecible.2,9,10 Existen reportes de casos en los cuales es útil el uso de toxina botulínica inyectada en los músculos cigomáticos para mioclonus facial y en el miembro superior en casos de epilepsia focal con el fin de disminuir los espasmos epilépticos, el dolor y mejorar la funcionalidad de la extremidad comprometida.2,11-13
Los reportes de series de casos han demostrado que en corto tiempo de seguimiento el efecto benéfico de la terapia inmunomoduladora, la plasmaféresis, los pulsos de esteroide y un ensayo clínico abierto con infusiones de rituximab como buena opción para disminuir la frecuencia de las crisis.14,15 En 2011 evaluaron el uso de estimulación del nervio vago y terapia trascraneal, pero muchos casos fueron refractarios al manejo.16,17
Un estudio reciente realizado por Bien y col. sobre el papel del tacrolimus como objetivo terapéutico sobre las células T y la respuesta inmunomediada, demostró una disminución en el deterioro funcional pero sin mejoría en la progresión de las convulsiones, sin embargo fue un medicamento mejor tolerado que la inmunoglobulina endovenosa.2,18
Otro estudio retrospectivo basado en las historias clínicas realizado por Caitlin E Hoffman y col. en el Hospital de Toronto entre 1983 a 2012 en 13 pacientes con diagnóstico clínico, imagenológico, electroencefalográfico e histopatológico de encefalitis de Rausmussen con edad media de 10 años, evaluó el efecto de la inmunoterapia, régimen antiepiléptico, progresión clínica, tiempo de tratamiento quirúrgico, morbilidad posterior a la cirugía, progresión clínica y resultado postoperatorio. Durante el seguimiento se observó que estas terapias aunque son una buena opción en pacientes no candidatos a terapia quirúrgica, no hay fuerte evidencia de reducción de las crisis, los efectos secundarios se asocian con incremento en la morbilidad y el resultado a largo plazo aún no ha sido demostrado; por lo tanto, la terapia temprana con hemisferectomía continúa siendo el patrón de oro en el manejo de la enfermedad, siendo esta una técnica usada desde 1950 pero su evidencia se limita a reportes de casos o estudios de cohortes en los cuales el tamaño de muestra es pequeño y aun no existen protocolos acerca de la mejor técnica quirúrgica.2,19,20 El caso reportado recibió tratamiento con inmunoglobulina endovenosa y en la actualidad recibe terapia combinada antiepiléptica con adecuado control de las crisis.
Conclusión
La encefalitis de Rasmussen es una patología de interés en la práctica médica que cursa con deterioro progresivo de funciones cognitivas y motoras en la población pediátrica y en menor proporción en adultos, lo que conlleva a discapacidad en edades tempranas con importante impacto social. Existen vacíos en la literatura acerca de la mejor opción terapéutica, dado que los estudios se limitan a series se casos y cohortes en los cuales se ha observado que la inmunoterapia, el uso de esteroides, plasmaféresis y rituximab utilizados como terapia de primera línea retardan la progresión pero no tienen un efecto sobre la frecuencia ictal ni disminuyen la posibilidad de hemisferectomía, siendo esta última la mejor opción de terapia a la fecha. Se requieren estudios adicionales con mejor metodología para resolver todos estos interrogantes.
RESPONSABILIDADES ÉTICAS
Protección de personas y animales: los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales. Confidencialidad de los datos: los autores declaran que han sido seguidos los protocolos de su centro. Derecho a la privacidad y consentimiento informado: los autores han obtenido el consentimiento informado por parte de padres responsables de la paciente menor de edad. Este documento obra en poder del autor de correspondencia.
CONFLICTO DE INTERESES
Los autores declaran no tener ningún conflicto de intereses.
Referencias